This is part of a series of stories on inflammation triggered by SARS-CoV-2 infection. Other articles in the series include:lectins, Covid-19 and brain injury, long Covid, STING, monocytes, and ORF8. They may also be found on my website, www.williamhaseltine.com.
When it comes to SARS-CoV-2 infection, our very own immune system can end up contributing to the damage. Inflammation, though an important part of defense against microbes, can sometimes get out of hand, leading to multiorgan injury and even death. This is evidenced by the hyper-inflammation commonly associated with severe Covid-19. The mechanisms by which SARS-CoV-2 triggers such an intense inflammatory response are still being uncovered. New work by a group of researchers at Peking Union Medical College adds another piece to the mosaic of causes. Published in Science Signaling, Liu et al. discovered that SARS-CoV-2 spike protein can prompt cells in the lungs to fuse together, causing damage to their DNA and triggering an immune response that culminates in excessive inflammation.
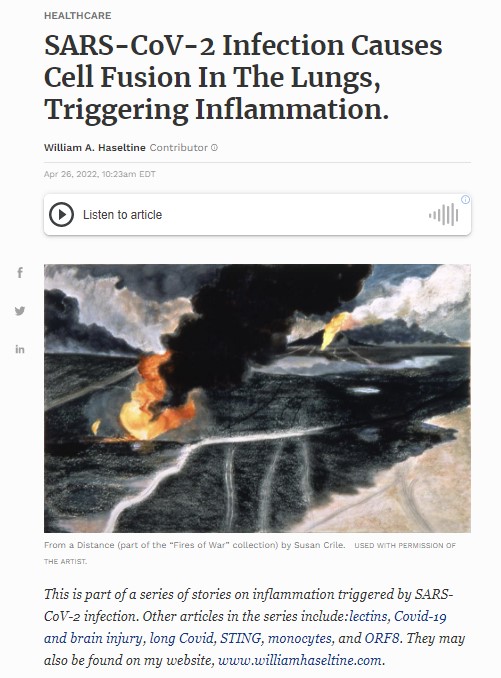
Although the impacts of SARS-CoV-2 infection have been recorded across the board —from the liver to the heart to the brain, and beyond— nowhere is the damage quite as intense as in the lungs. The presence of syncytia, along with alveolar destruction and massive blood clotting, are all characteristic of severe Covid-19. Syncytia are large, cell-like structures that form when two or more cells join together; essentially, an amorphous mass of cytoplasm and nuclei (Figure 1). Even though syncytia are crucial to the formation of our muscle tissue, when induced in response to viral infection they are pathological. Importantly, the syncytia formed in response to SARS-CoV-2 infection do not seem to heal or regenerate with time, as they do in response to the flu.
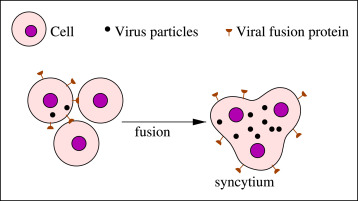
To study the formation of syncytia and its knock-on effects, Liu et al. co-cultured embryonic kidney cells containing SARS-CoV-2 spike protein and enhanced green fluorescent protein (EGFP) —which makes the S protein-containing cells show up green under a microscope— with a range of ACE2-containing human endothelial cells. ACE2 acts as the primary portal of entry for SARS-CoV-2, mediated by the spike protein.As expected, the researchers witnessed cell fusion between the spike protein-containing cells and the endothelial cells. This had already been demonstrated in prior studies. But Liu and colleagues noticed that cell fusion depends greatly on host proteases used to break down the spike protein, such as transmembrane serine protease 2 (TMPRSS2). SARS-CoV-2 spike undergoes cleavage at two sites: S1/S2 and S2’ (Figure 2). When the researchers generated spike protein mutants with alterations to these sites —rendering them resistant to cleavage— they witnessed only modest, if any, syncytia formation. The unmutated spike protein, in contrast, induced extensive cell fusion.
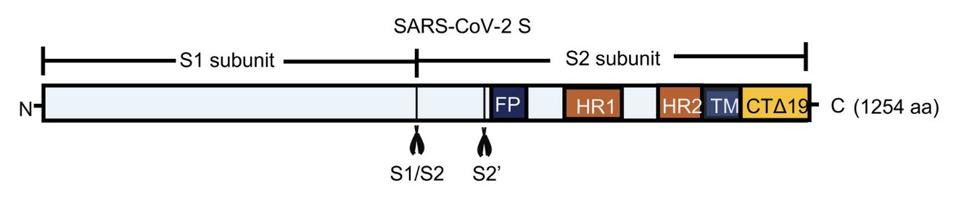
FIGURE 2. Schematic representation of the S1/S2 and S2’ cleavage sites along the SARS-CoV-2 spike … [+]FROM: “SARS-COV-2 SPIKE PROTEIN–INDUCED CELL FUSION ACTIVATES THE CGAS-STING PATHWAY AND THE INTERFERON RESPONSE” LIU ET AL. 2022
Following cell fusion and the formation of syncytia, Liu et al. witnessed upregulation of interferon-beta (IFN-β). Interferons are a class of signaling proteins produced and released by host cells in response to viral infection, warning other nearby cells of danger. IFN-β is particularly closely linked to the innate immune response and, crucially, the activation of inflammation. Although helpful in containing viral spread across the body, prolonged and misregulated production of interferons can impair lung cell regeneration and worsen disease outcome.
Curious to determine how exactly cell fusion triggers IFN-β activation, the team of scientists turned to one of the central signaling pathways of the innate immune system, the cGAS–STING pathway (Figure 3). This pathway kicks off with the detection of misplaced DNA by an enzyme called cGAMP synthase (cGAS). DNA is usually found in the nucleus of a cell, so its presence in the cytoplasm indicates either cellular damage or microbial infection. cGAS binds to the misplaced DNA, triggering the formation of cyclic GMP-AMP (cGAMP). This, in turn, goes on to bind to Stimulator of Interferon Genes (STING), a protein in charge of inducing the production of type 1 interferons such as IFN-β. With STING in the game, interferon regulatory factor 3 (IFR3) undergoes phosphorylation. Once phosphorylated, IFR3 can travel to the nucleus of the cell in order to upregulate inflammatory genes.
FIGURE 3. A simple diagram illustrating the various steps involved in the cGAS–STING pathway, … [+]SOURCE: BELLBROOK LABS
Liu et al. returned to their co-cultured cells and looked for signs of cGAS–STING pathway activation. Substantiating their hunch, they found traces of phosphorylated IRF3 in the cell culture. To further confirm their hypothesis, they co-cultured cells engineered to lack cGAS and STING with spike-protein containing cells. The result? A marked decrease in IFN-β activation.
Interestingly, the researchers found that stimulation of IFN-β also depended on spike cleavage. Returning to the cleavage-resistant spike protein they had engineered, Liu et al. compared the activation of the cGAS–STING pathway between the mutated spike and the original SARS-CoV-2 spike. Only the unmutated spike protein induced the expression of IFN-β. They also found that cells overexpressing TMPRSS2 displayed higher levels of IFN-β expression.
So the cGAS–STING pathway is responsible for triggering IFN-β upregulation in response to SARS-CoV-2-induced cell fusion. But, how is cell fusion triggering the pathway?
Genetic analysis of the cell cultures that triggered the cGAS–STING pathway revealed an increased induction of DNA damage responses (DDR) — an elaborate network of genes in charge of recognizing and tending to DNA damage. Liu et al. turned to the microscope; they observed that spike protein-induced syncytia were peppered with ruptured nuclei and with micronuclei. This was not seen in co-cultures formed using the cleavage-resistant, mutated spike protein.
Finally, the researchers sought to confirm whether the same series of events unfold during viral infection. Instead of the spike protein-containing kidney cells they used for their initial co-cultures, the team directly infected a range of human endothelial cell lines with a vesicular stomatitis virus (VSV) that had been engineered to express the SARS-CoV-2 spike protein. Again they witnessed the formation of syncytia, ruptured nuclei, the activation of the cGAS–STING pathway, and ultimately, the upregulation of IFN-β.The work by Liu et al. offers us insight into another mechanism through which SARS-CoV-2 induces inflammation. Covid-19 patients initially display a weakened type 1 interferon response, in large part orchestrated by the virus’ immunosuppressive tactics. This isn’t the case with patients that progress to severe disease, who often display signs of robust interferon response. More research needs to be done to understand the contribution of type 1 interferon to hyper-inflammation and severe Covid-19.