This is part of a series of stories on inflammation triggered by SARS-CoV-2 infection. Other articles in the series include: lectins, Covid-19 and brain injury, and long Covid. They may also be found on my website, www.williamhaseltine.com/.
Inflammation is a key feature of SARS-CoV-2 infection, both during the acute phase of Covid-19 and also as a contributor to post-acute sequelae SARS-CoV-2 infection (PASC), otherwise known as long Covid. Inflammation in reaction to any microbe is Janus-faced. On the one hand, it can serve as a barrier to prevent the spread of, and also destroy, invading pathogens. On the other hand, if uncontrolled, inflammation can lead to serious multi-organ damage and even death. New research from Harvard Medical School and Boston Children’s Hospital provides insights into the multiple ways SARS-CoV-2 can trigger inflammation. Junqueira et al. found that non-productive infection of monocytes and macrophages stimulates the release of potent inflammatory signals, explaining at least in part the inflammation associated with Covid-19. The study was based on a comparison of blood and lung tissues from a matched-set of 22 Covi-19 patients and 19 healthy donors.
The researchers searched for markers of pyroptosis, a form of programmed cell death. Cells are equipped with multiple alarms that signal the presence of an invading microorganism or inappropriate cell damage. These alarms are set of either by pathogen-associated molecular patterns (PAMPs) — molecular “motifs” that are preserved across most pathogens, be they viral, bacterial, or fungal— or by damage-associated molecular patterns (DAMPs), released by damaged or dying host cells. Some of these alarms stimulate the inflammatory response, mediated by a protein signaling complex dubbed the inflammasome. Once assembled, the inflammasome activates a protease called caspase-1. This, in turn, stimulates the production of proinflammatory cytokines, like interleukin-18, and a pore-forming protein called Gasdermin-D. Gasdermin-D punches holes in the cell’s membrane, eventually causing it to explode and spill all of its contents. This helps call other immune cells to the area, increasing inflammation and assisting with host defense. It also prevents the host cell from having its cellular machinery hijacked by the pathogen for purposes of self-replication. The full pathway leading to pyroptotic cell death is depicted in Figure 1.
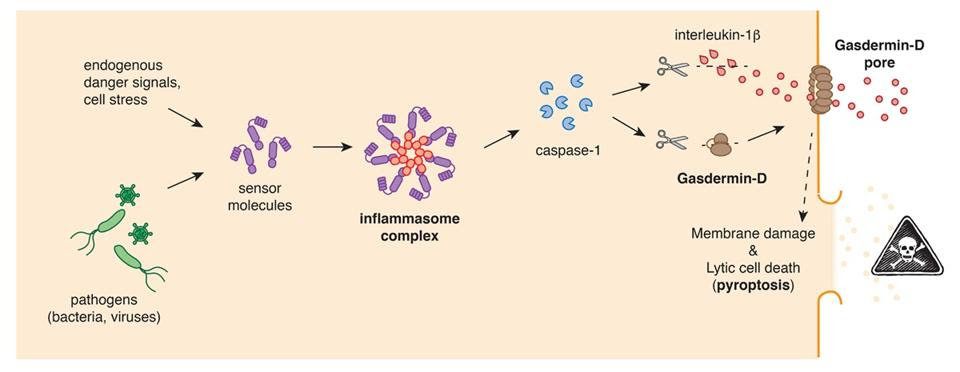
But sometimes this process goes haywire and fails to reel itself in, causing extreme inflammation that actually furthers tissue and organ damage.The researchers discovered that the cells showing the most signs of pyroptosis —as determined by cell membrane damage and inflammasome activation— were monocytes. These are immune cells that act as lookouts, keeping an eye out for any microbes in the bloodstream, and also serve as early responders to infection. Lung macrophages, a similar immune cell found in tissue instead of the blood, also showed signs of pyroptosis.
“In the infected patients, about 6 percent of blood monocytes were dying an inflammatory death,” said Judy Lieberman, senior author of the study. “That’s a large number to find, because dying cells are rapidly eliminated from the body.”
Junqueira et al. then investigated the possibility that the monocytes and macrophages themselves were being infected by SARS-CoV-2. To do so, they engineered a SARS-CoV-2 clone that encoded a fluorescent reporter of viral replication. They found that about 10% of monocytes and 8% of lung macrophages displayed clear signs of infection. This included the presence of viral RNA and viral proteins. No such monocyte or macrophage infection was seen in the control group of healthy blood donors.
Curiously, the researchers noticed that lung epithelial and endothelial cells, whose surfaces are covered with angiotensin converting enzyme 2 (ACE2) receptors, the primary portal of entry for SARS-CoV-2 infection of cells, showed neither signs of inflammasome activation nor signs of pyroptosis.
Monocyte infection was unexpected as the well-known ACE2 receptor is absent from the cell’s surface, and found only in very low concentrations on the surface of macrophages. But they were clearly being infected. So, how is SARS-CoV-2 binding to and entering these cells?
The team of researchers suspected that infection may be occurring via antibody-dependent enhancement (ADE). Antibodies are produced by our adaptive immune system in response to microbial threat. Although there are a variety of different antibodies, they generally fall into one of three functional categories: those that bind to the surface of a pathogen to prevent it from being able to enter its target cell (neutralizing antibodies); those that cover the surface of a pathogen and mark it for removal by monocytes, macrophages, and other immune cells (opsonins); and finally, those that trigger other immune responses, including the complement pathway, to help eliminate pathogens. Put simply, when non-neutralizing antibodies bind to a pathogen they sometimes facilitate the pathogen’s entry into cells that carry antibody receptors (Figure 2).
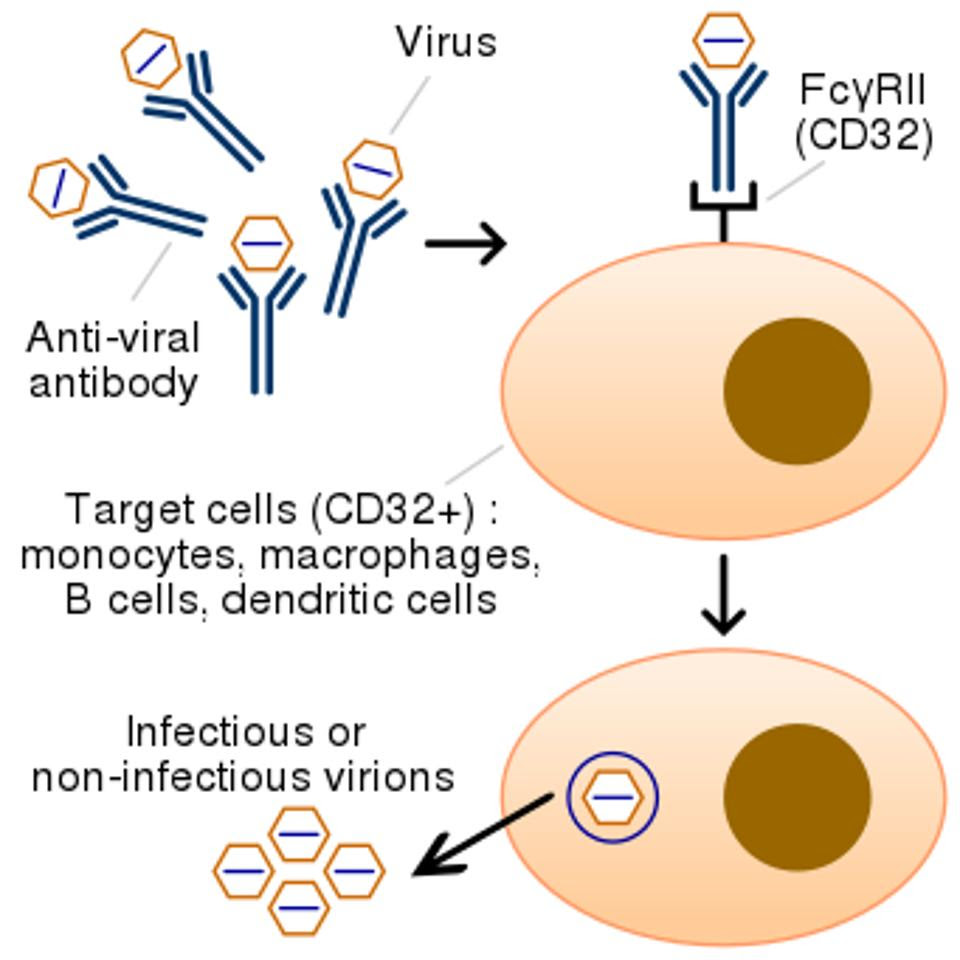
Junqueira et al. discovered that a particular subset of monocytes were especially sensitive to SARS-CoV-2 infection: those that express the CD16 receptor. This is one of three receptors monocytes use to bind with antibodies. Although CD16 monocytes usually comprise only 10% of the total monocyte population, they are noticeably increased in Covid-19 patients.
Confirming the team’s suspicions, CD16 receptors were able to recognize viral particles tagged by antibodies, allowing them access into the cell. This did not happen with monocytes from the healthy donor group, unless mixed with anti-Spike antibodies or antibody-containing plasma from Covid-19 patients.
Crucially, vaccine-derived antibodies did not facilitate viral entry into monocytes. Although not entirely sure why this is, Junqueira et al. hypothesize it may come down to differences in the properties of the antibodies, with those developed post-vaccination less prone to binding to CD16.
This work provides clear evidence that SARS-CoV-2 infection of monocytes and macrophages does occur and that, when it occurs, it triggers an inflammatory response. However, the researchers noted that infection of the monocytes and macrophages does not lead to the production of infectious virus particles. Evidently the full replication cycle is blocked, possibly by the inflammatory response itself.
“In some ways, uptake of the virus by these ‘sentinel’ cells is protective: it sops up the virus and recruits more immune cells,” said Lieberman. “But the bad news is that all these inflammatory molecules get released. In people who are more prone to inflammation, such as the elderly, this can get out of control.”
Junqueira et al.’s research goes partway to explain the inflammatory response of those infected with SARS-CoV-2. Other contributors include lectins, which distort our neutrophils, and innate lymphoid cells (ILCs), which produce proinflammatory chemokines like interleukin-17. Clearly, unless infection is controlled by the initial innate immune response, or high-levels of neutralizing antibodies, virus replication can trigger a violent cycle of hyper-inflammation leading to severe disease and even death.