Systemic lupus erythematosus (SLE), more commonly known as simply lupus, is a chronic autoimmune disease that provokes symptoms from skin rashes and fevers all the way to chronic fatigue and inflammation of joints and organs. Severe cases of lupus can be fatal. Autoimmune diseases occur, in the broadest sense, when our body’s natural defense, the immune system, mistakes our own cells or tissues for foreign substances, accidentally turning on us and attacking otherwise healthy cells and tissues — in essence, a lapse in the ability to tell self from other. Even so, at a more granular level, the exact causes and mechanisms of the self-targeting seen in autoimmune diseases remain poorly understood. It is likely that a complex mix of genetic and environmental factors come together to trigger disease onset. A new study by researchers at the Australian National University, Canberra heralds a major breakthrough. Published in Nature, the work by Brown et al. pinpoints a mutation in the gene of a protein that senses single-stranded RNA as one direct cause of systemic lupus erythematosus. The same protein, toll-like receptor 7 (TLR7), is activated upon infection by single-stranded RNA viruses like SARS-CoV-2.
Toll-like Receptors (TLRs): What are they and what do they do?
Toll-like receptors are a family of proteins closely involved in the innate immune response. They are usually found in the membrane of sentinel cells —macrophages and dendritic cells— that circulate throughout our body while keeping an eye out for pathogens. These cells are often the first to respond to infection, and it’s the toll-like receptors in their membrane that help them to spot any possible microbial threats. As pattern recognition receptors, toll-like receptors can detect molecular “motifs” that are highly conserved across pathogens, from bacteria and viruses to fungi and parasites. These motifs are known as pathogen-associated molecular patterns (PAMPs). When toll-like receptors uncover traces of PAMPs in places they shouldn’t be, they quickly activate and mobilize different immune cells.
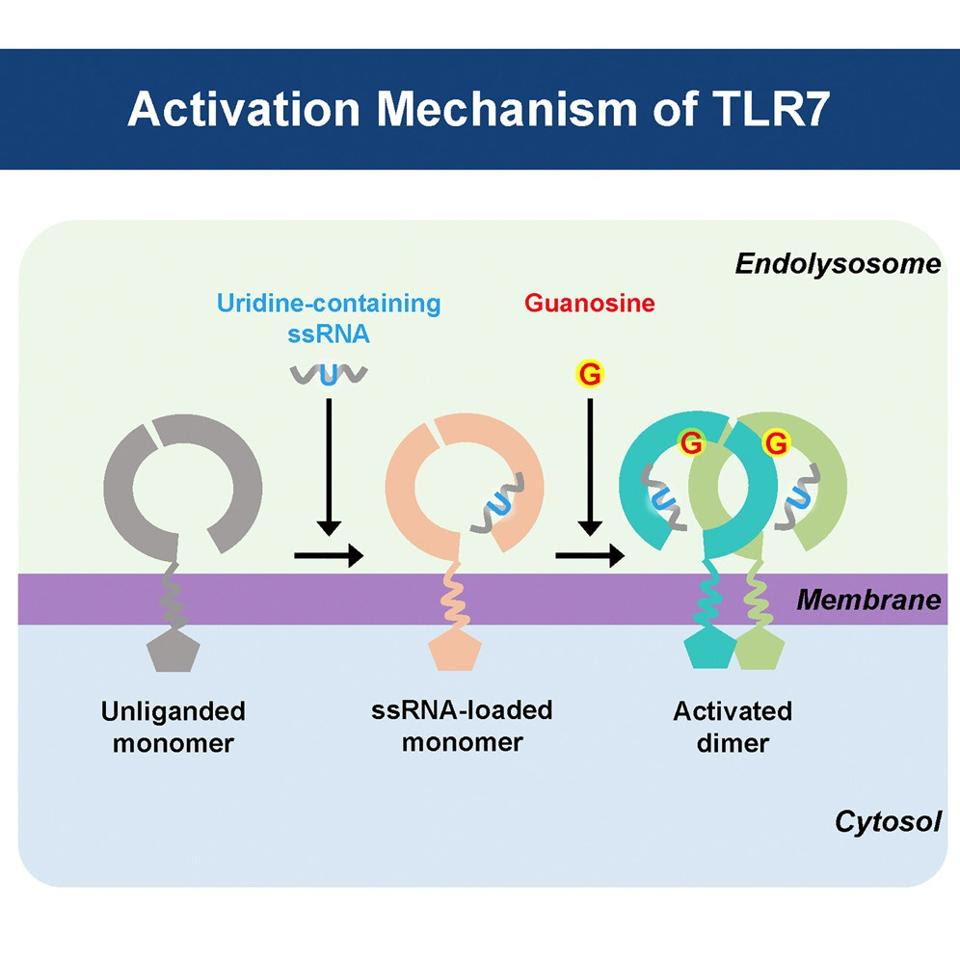
Toll-like receptor 7, one of the 10 different types found in humans, is in charge of detecting single-stranded RNA in the endosomes of cells — endosomes are “sorting stations”, located within the cytoplasm of cells, that help send important proteins to various destinations within the cell. Single-stranded RNA is a hallmark of certain viruses, including hepatitis C virus, human immunodeficiency virus (HIV), and coronaviruses such as SARS-CoV-2. TLR7 is particularly responsive to guanosine and uridine, two nucleotides derived from single-stranded RNA (Figure 1).
Once TLR7 detects viral single-stranded RNA, it stimulates a series of signaling cascades that ultimately activate nuclear factor-kappaB (NF-κB). NF-κB is a protein complex that regulates a host of important cellular behaviors, including: the induction of type I interferon (INF1) and other inflammatory proteins, like cytokines; cellular growth; and, finally, the programmed self-destruction of cells, known as apoptosis. NF-κB is especially closely involved in the regulation of B lymphocyte survival, preventing apoptosis and promoting activation whenever possible.
TLR7, Autoimmunity, and Lupus
As mentioned, autoimmune diseases are a product of our immune system mistakenly attacking our own healthy cells and tissues, rather than a foreign threat. For the most part, the things that do the attacking are antibodies, which are secreted by B cells. Since they end up directed at us instead of microbial threats, they are called autoantibodies. The cells and tissue they end up attacking are called autoantigens.
Although an important component of the antiviral immune response, TLR7 has also been associated with autoimmune disorders. This is in large part because of its role in regulating inflammation and B cell survival via NF-κB. In the case of lupus, it has long been suspected that increased TLR7 signaling may be a major contributor. This new research by Brown et al. confirms the suspicion.
To study the relationship between lupus and TLR7, the team of scientists performed a full genome analysis of a young Spanish woman who was diagnosed with systemic lupus erythematosus at the age of 7. Such early onset is quite unusual, and hints at a distinct genetic cause. Indeed, the genetic analysis revealed that she had a mutation to the TLR7 gene, a switch from tyrosine to histidine, giving rise to a new variant: TLR7Y264H. The researchers cross-referenced the mutation against databases of normal human genome variation, but didn’t get any results.
Next, Brown et al. sought to check if this variation enhances TLR7 signaling and the related downstream effects, like NF-κB activation. To do so, they engineered different cells to express the three TLR7 mutants they had discovered. They then activated the variants and measured the level of TLR7 expression. Compared to the unmutated TLR7 gene, TLR7Y264H expressed higher levels of the TLR7 protein than usual and induced enhanced NF-κB activation.
The TLR7Y264H gene variant also increases the protein’s affinity to the activator guanosine, which dimerizes and activates the receptor (Figure 1). This increased sensitivity of TLR7 makes it more likely for our immune cells to mischaracterize our own healthy tissues as either damaged or as a foreign substance, causing them to go on the offensive.
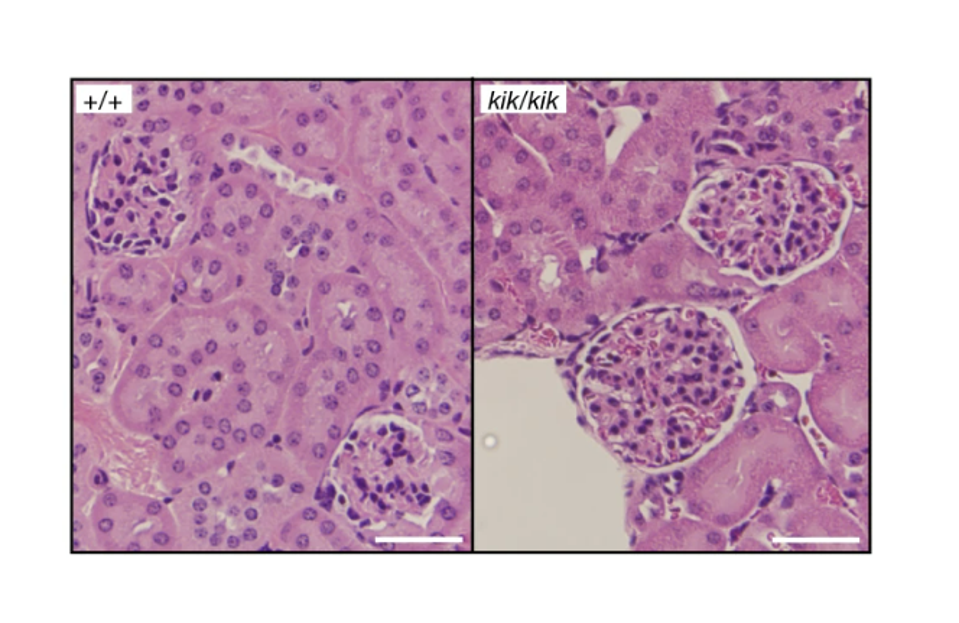
To see what this meant in the context of autoimmunity, the team of scientists introduced the Y264H mutation into mice. Both male and female mice carrying the TLR7 variant began displaying signs of autoimmune symptoms within 12 weeks. This included tissue damage at various different sites and an overall decreased survival compared to unmutated mice (Figure 2). The symptoms were accompanied by the development of autoantibodies towards single-stranded RNA and Smith proteins (Sm), a family of RNA-binding proteins that help splice protein-producing sections of RNA. Presence of autoantibodies against Smith proteins has been used as a diagnostic criteria for lupus. So, along with the symptoms, the presence of these autoantibodies suggested that the TLR7Y264H variant was directly responsible for the onset of lupus in the mice.
The researchers then focused on the impact the Y264H variant of TLR7 had on B cell production and survival. Analysis of the spleens of mice with the TLR7Y264H variant showed an increase in total B cells as compared to unmutated mice. B cells derived from the mutated mice also survived much longer, with a two-fold increase in the activation of genes related to cell survival. Their results suggest that the hypersensitive TLR7Y264H variant promotes the survival of autoantibody-producing B cells.
Importantly, Brown et al. were able to confirm that the effects of the TLR7Y264H variant were independent of germinal centers (GCs). When B cells encounter an antigen they travel to germinal centers, where they start developing specific antibodies to neutralize the substance. One critical function of the germinal centers is to filter out those B cells that fail to differentiate self from other. B cells that have never been filtered through a germinal center are much more likely to accidentally start targeting our own healthy cells and tissues. Introducing the Y264H mutation into mice that cannot form germinal centers still causes lupus, suggesting that the autoimmunity is being driven by TLR7 independently of germinal centers.
Implications for Long Covid?
There appear to be some parallels between the TLR7-driven autoimmunity seen in lupus and the autoimmune aspects of post-acute sequelae SARS-CoV-2 infection (PASC), also known as long Covid.
First, being a single-stranded RNA virus, SARS-CoV-2 also induces TLR7 activation. This has been confirmed by studies showing that individuals with TLR7 deficiencies are more likely to develop severe Covid-19. Second, like lupus, SARS-CoV-2 infection is accompanied by a proliferation of B cells that have not passed through germinal centers, and therefore maintain the capacity for self-recognition. Both lupus and long Covid are characterized by the persistence of such auto/antibodies. Also, both lupus and long Covid are more prevalent in women than in men. Given the results of their latest study, Brown et al. suggest that, in lupus, this may be because the TLR7 gene is found on the X chromosome, and females have two X chromosomes where males have one X and one Y chromosome. Although one X chromosome is usually inactive in women, silencing of the section of the chromosome on which the TLR7 gene sits is often incomplete.
Dr. Carola Vinuesa, senior author of the study, mentions that, “There are other systemic autoimmune diseases, like rheumatoid arthritis and dermatomyositis, which fit within the same broad family as lupus. TLR7 may also play a role in these conditions.” Perhaps TLR7 may also be to blame for the autoimmune aspects of long Covid? At the very least, the many parallels between the two suggest it may be worth taking a closer look.