Once again, Covid-19 is on the march around the world. Most infections are due to the Delta variant ravaging the Indian subcontinent in the spring and early summer. A second variant, Lambda, is wreaking similar havoc in South America.
The Lambda variant, also known as “C.37,” is driving a rapid increase of Covid-19 throughout South America, Central America, and parts of the Caribbean. First detected in Peru as early as August 2020, the variant is now present in over 30 countries and is now the dominant strain in various Latin American countries.
Early studies by the University of Chile indicate that the variant confers significant resistance to neutralizing antibodies induced by the inactivated CoronaVac vaccine, one of the major vaccines used in South America. The study showed a 3-fold drop in neutralization against the Lambda variant relative to the wild-type virus, compared to a 2.3-fold drop for the Gamma variant, and a 2-fold drop for the Alpha variant. The effectiveness of the mRNA vaccines the variants has yet to be reported.
The Lambda Genome
Figure 1 illustrates the nucleotide mutations and related amino acid substitutions or deletions of Lamba versus the canonical Wuhan strain.

FIGURE 1: Common mutations external to the S protein in the lambda variant genome. Color code: noncoding nucleotide mutations (red), synonymous mutations (blue), nonsynonymous mutations (pink), and their corresponding amino acid mutations (purple).
Beginning at the 5′ end of the genome, we note C241U in the 5′ untranslated terminal region (5′ UTR). The 5′ UTR change, as well as NSP12-P323L and S-D614G, are present in all G-clade variants. The G-clade arose in February 2020. It is now the rootstock of most variants of interest or concern. The D614G mutation in the S protein is critical to increased transmissibility. The contributions of the 5′ UTR and NSP12 mutations to increased infectivity are speculative.
Lambda contains six common mutations in the NSP3 gene, three of which are silent and three that change the protein sequence. NSP3 is a complex seven domain protein essential to the establishment of productive infection. NSP3 contributes to creating the double-membrane vesicle, which shields the replication-transcription complex from recognition by the innate inane system. NSP3 also encodes de-ubiquinase and de-ADP-ribosylation activities. Additionally, NSP3 comprises the pore through which viral genome and mRNA enter the cytoplasm. We note that two of the amino acid changes, P1469S and F1569V, lie in transmembrane domains. The NSP4 protein also participates in the formation of the replication-transcription double-membrane vesicle. Both of the mutations in NSP4, L438P, and T492I, also lie in transmembrane domains. NSP6, the third protein involved in creating the double-membrane vesicle, likewise contains a three amino acid mutation that alters a transmembrane domain. This observation underscores the relevance of these mutations to the biology of SARS-CoV-2 that one of these changes, L438P, is found in the Iota variant, which arose independently in New York. The contribution of changes in these three proteins is worthy of study to clarify their potential role in increasing the replication competence of the Lambda variant.
The S protein of Lambda harbors eight common mutations, one of which is synonymous. Six of the remaining mutations, three in the amino-terminal domain (NTD) and two in the receptor-binding domain, and one in the amino terminus of the S2 protein, are unique to the Lambda variant. (Figure 2). The three NTD mutations likely contribute to escape from neutralizing antibodies, as do mutations in the NTD of other variants.
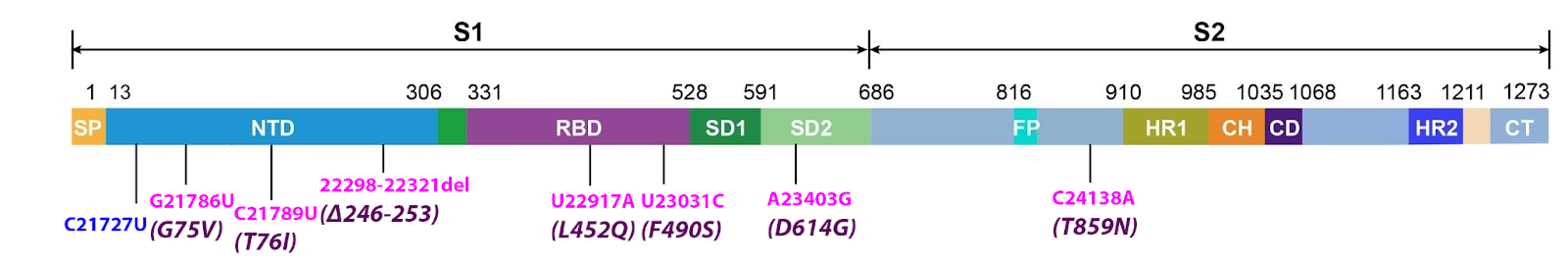
FIGURE 2: Common mutations to the S protein in the lambda variant genome. Color code: synonymous nucleotide mutations (blue), nonsynonymous nucleotide mutations (pink), and their corresponding amino acid mutations (purple).
The L452Q mutation is the receptor-binding domain that results in the substitution of leucine for glutamine. This is a change from a hydrophobic residue to a hydrophilic and polar one. The effect of this change is likely to be similar to the L452R (leucine to arginine) mutation found in other armed variants, such as the delta variant and the B.1.427 and B.1.429 strains first identified in California. As reported for the L452R mutation, this change likely increases the avidity and decreases neutralizing antibody recognition of the S protein.
The S protein also includes the amino acid change F490S. The F490S mutation is unique to Lambda among major variants. However, identical mutations arose in two in vitro experiments. Schreiber et al. searched for mutations that increased the avidity of the S protein for the ACE2 receptor. An independent set of experiments mapped S proteins mutations that contribute to escape from neutralization by convalescent antisera. The F490S change likely contributes to both properties of the Lambda variant.
The only mutation of the 3′ Orf proteins occurs in Orf9b, P10S. Orf9b plays a central role in the suppression of the innate immune system, inhibiting the mitochondrial antiviral signaling protein (MAVS), the TNF receptor-associated factor 3 (TRAF3), and TRAF6. Over-expression of Orf9b is characteristic of the Alpha variant. The possibility that this mutation increases the activity of Orf9b deserves attention.
The nucleocapsid protein (N) contains five mutations, all of which change amino acids. N is a multifunctional protein. The core of the virus particle is an N protein-virus genome complex. Early in infection, the N protein suppresses innate immunity by antagonizing interferon beta, inhibiting cyclin-cyclin-dependent kinase, and inhibiting nonsense-mediated decay. Late in infection, the N protein is reported to contribute to hyper-immune activation. Three of the mutations (P13L, G214C, and T366I) are unique to the Lambda variant. Many variants of concern and interest (Alpha, Gamma, B.1.1.298, and B.1.1.28.3) harbor mutations identical to R203K and G204K. The contributions of N to the properties of variants are well worth additional investigation.
Researchers generally ignore synonymous nucleotide changes when describing the properties of variants. However, a recent study shows that nucleic acid changes in the genome contribute to the success of variants. Thorne et al. describe what appear to be cis-acting nucleotide changes that contribute to an astonishing 80 fold increase in the N/orf9b messenger RNA. A complete account of the properties of variants may well require an understanding of the contributions of mutations that affect both protein and non-protein-coding genome variants.
The figure below shows an aggregation of mutations throughout the genome found both in Lambda and other variants of interest and concern as described by the World Health Organization (Figure 3).

FIGURE 3: Mutations in the lambda variant genome that appear in other variants of interest and concern as described by the World Health Organization.
We are just beginning to understand the full complexity of Covid-19 as a pandemic and as a disease. To understand both, we must understand SARS-CoV-2, the virus that lies the source of our troubles. This is a pathogen that has successfully evolved over millions of years to infect populations much like ours. In order to survive, the virus must infect and reinfect animals with well-honed innate and adaptive immune systems. It is naive to assume that changes in the spike protein alone account for all the properties of variants that afflict us today. I am convinced that each new variant provides a clue to the virus’s success, ones we can exploit as weaknesses if we only knew enough. Fortunately, we have the tools and the experts that are more than up to the task.