One of the major time hindrances to the development of monoclonal antibodies is one of the first steps: identification. Advances in artificial intelligence antibody modeling may pave the way to reduce time spent during the identification process. Rather than sorting through millions of B-cell receptor sequences manually or with the assistance of some software, what if that work could be expedited by artificial intelligence?
There’s an urgent need for drugs to prevent and treat Covid-19, particularly for immunosuppressed people. Unfortunately, the current Covid-19 variants have mutated to escape our currently approved antibodies. There are, however, a series of broadly neutralizing antibodies in research and development, though these typically suffer with lower affinity than would be ideal. Fortunately, a recent paper outlines a solution to this conundrum: using artificial intelligence to dramatically increase the binding affinity of antibodies.
Researchers Parkinson et al. present an artificial intelligence pipeline, RESP, which efficiently and independently identifies high-affinity antibody candidates. RESP selects the best fit to improve it for existing antibodies, given a particular antibody target. Using RESP, the researchers demonstrate an increased affinity to an existing drug by almost 20-fold. Here we examine the function of RESP and how it could improve antibody development in the near future.
RESP Components
The researchers describe the artificial intelligence system in four components. The first is an encoding scheme to pick out human B-cell receptor sequences from a larger panel. B-cells carry the genetic sequence used to encode and produce antibodies. By immediately recognizing human B-cell receptor sequences, RESP can immediately filter out innumerable sequences the researchers are not interested in.
The second component is a yeast surface display library to demonstrate the relative impact of mutations in an identified antibody’s sequence. Modest mutations of just one amino acid could radically impact a given antibody’s binding and neutralization capabilities. The sorted library can then be easily analyzed for different factors such as off-rate, half-life, binding affinity, and so on.
The third component is a sorting model for understanding the potential affinity of an antibody. RESP analyzes the off-rate of predicted antibody candidates and selects the highest affinity. Off-rate is essentially a ligand-protein binding component that negatively correlates with affinity. The lower the off-rate, the higher the affinity. They note this model can be retrained for different antigens, expanding the usefulness of RESP even further.
The fourth component aggregates the data from the preceding three to predict B-cell sequences with significantly lower off-rates, yielding antibodies with significantly higher affinity. Altogether, RESP seems impressive, but does the data back it up?
RESP Testing
The first test the researchers gave to RESP was for reconstructing sequences and discarding decoy sequences. The researchers introduced over 2.7 million human B-cell receptor sequences to RESP, as well as another 2.7 million decoys that had been modified in several positions. The system reconstructed the 5.4 million sequences with >99.99% accuracy, while its identification of decoys was a more modest but still strong 97.4%.
They next tested the validity of the yeast surface display library. Using approved monoclonal antibody treatment Atezolizumab, a cancer immunotherapy treatment for non-small cell lung cancer, they scanned for potential modifications in the antibody coding sequence that could improve the off-rate. Upon examining the heavy chains of the antibodies for positive mutations, the yeast library revealed over 92,000 unique sequences that were at least equal to the off-rate of the original Atezolizumab, many of which were lower, which we will speak to in a moment.
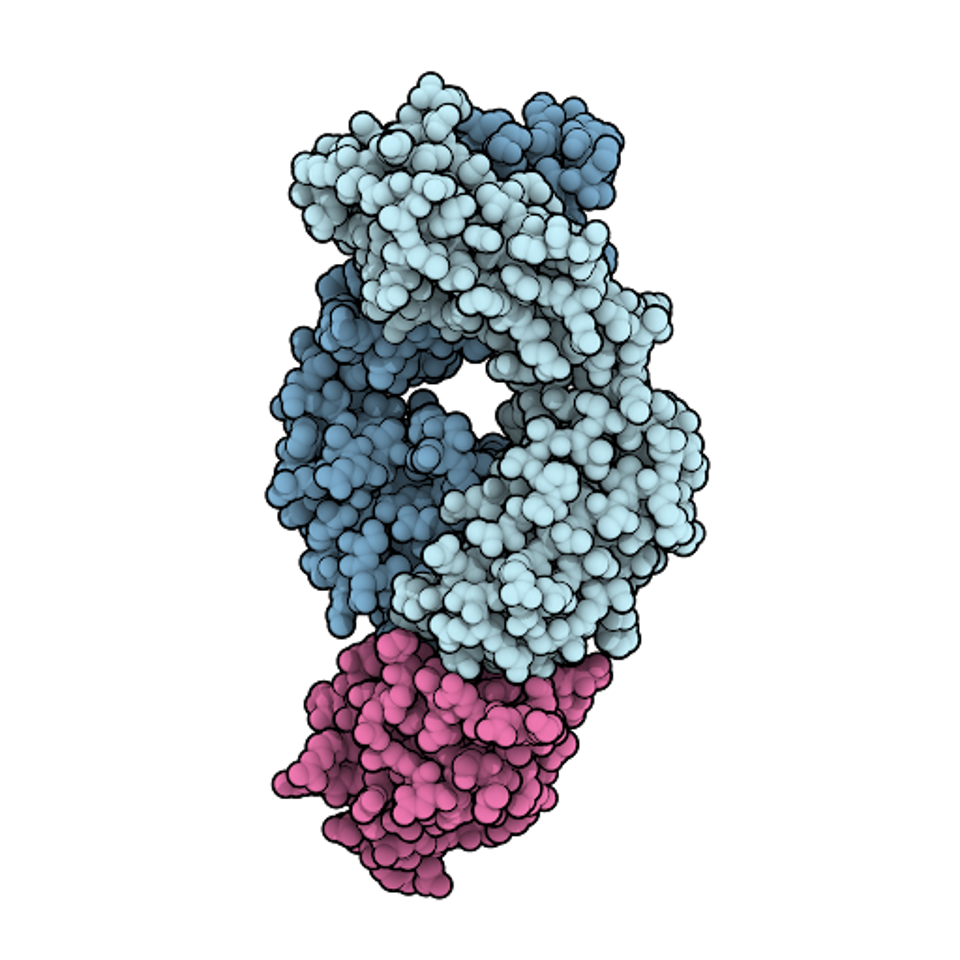
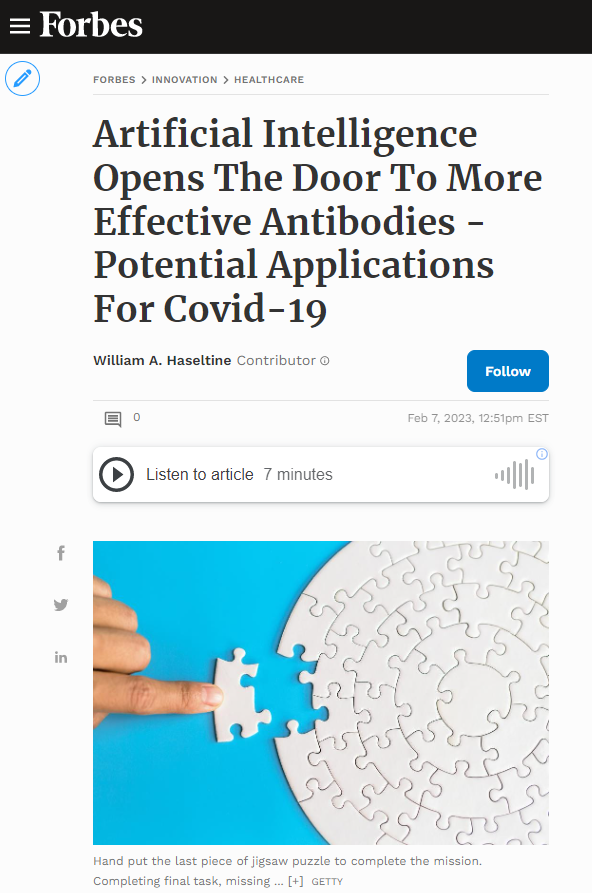
FIGURE 1: Space-filling model of the antigen-binding fragment of atezolizumab (pale blue) in complex with PD-L1 (pink).
DAVID GOODSELL – SCRIPPS RESEARCH INSTITUTE
Using the reconstruction and yeast library functions, RESP identified 21 final candidates with higher affinity than the original Atezolizumab. The 21 sequences carry mutations at residues A40, K43, T58, I70, N77, A79, S85, A97, and R98 in various combinations. None of these mutations appear in the original Atezolizumab and likely account for the increased binding affinity of the new candidates.
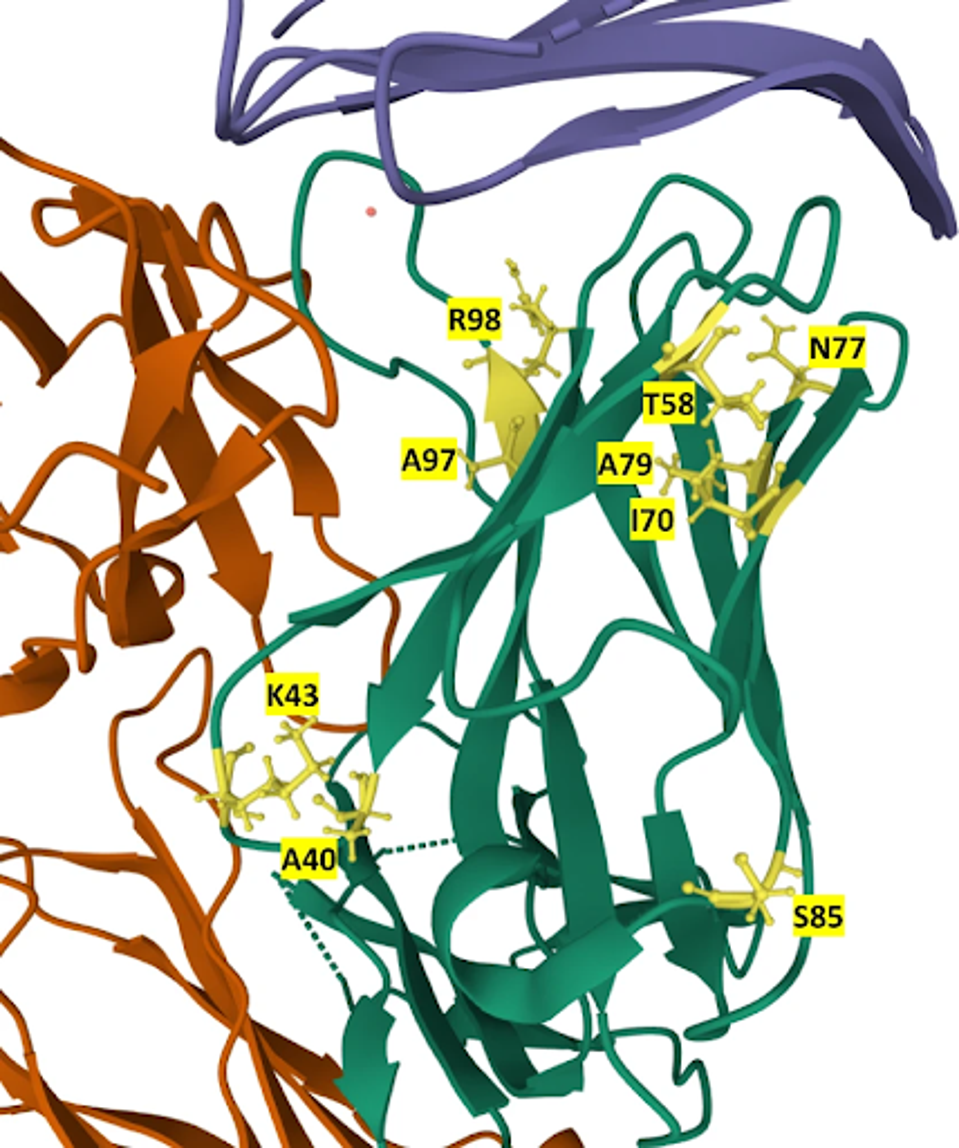
FIGURE 2: Location of mutations in the 21 mutants in the structure of the Atezolizumab heavy chain. Mutated residues are labeled and colored yellow in the heavy chain (green). PD-L1 is colored blue, while the light chain is orange.
PARKINSON ET AL.
One of the 21, mutant 4, carried mutations I70A, A79T, and A97V. Mutant four displayed significantly slower off-rates as compared to the unaltered Atezolizumab. And finally, this selected mutant bound the antigen 17-fold tighter than the approved drug.
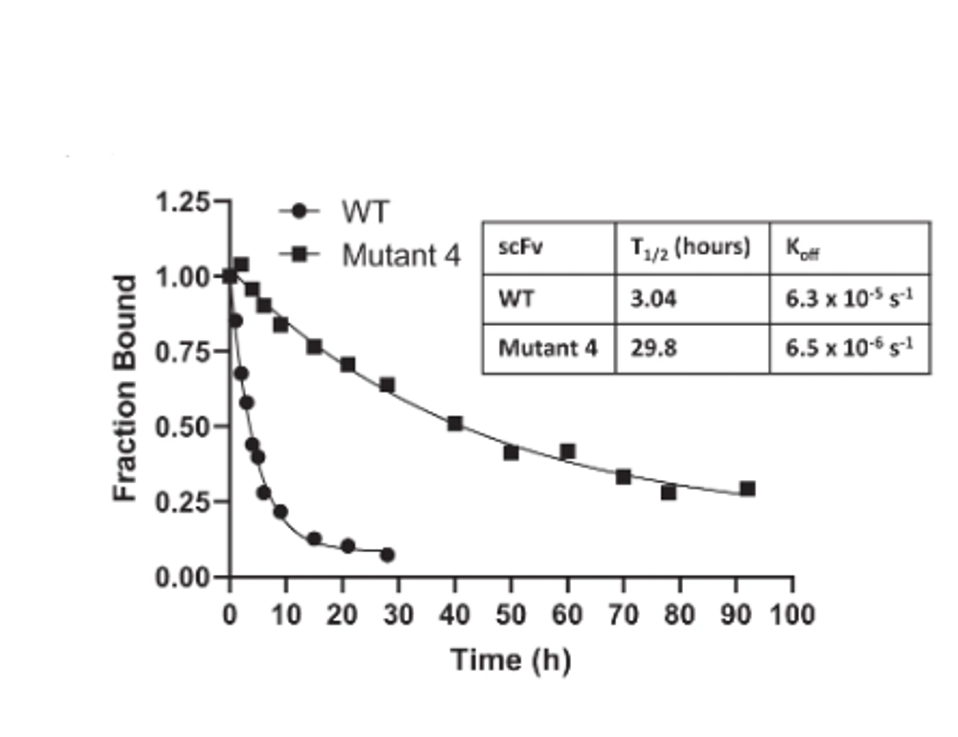
FIGURE 3: WT vs. Mutant 4 off-rate comparison.
PARKINSON ET AL.
Discussion
As with many aspects of our lives, artificial intelligence presents an opportunity to reduce manual input significantly and significantly improve productive output in antibody discovery. The most important element of RESP is its adaptability. This is not a Covid-19 antibody discovery mechanism or a mechanism for this specific version of cancer. RESP can be modestly altered to discover new or improved antibody candidates for all antigens, potentially even those without treatment.
If the trial run of RESP can identify a mutated alternative to an approved drug with a 17-fold increase in binding affinity, it could do the same for existing Covid-19 medicines and drug candidates. While further testing should always be pursued, the advancement of RESP should be pursued with haste.
Artificial intelligence mechanisms like RESP should be applied to the in-development panel of monoclonal antibodies. While previously and currently approved antibodies typically bind the receptor-binding domain of SARS-CoV-2, that is not the only target. For instance, the CV3-25 antibody described by Li et al. binds the S2 region of the virus on the complete other end of the spike. Another is the COV44-62/79 antibodies described by Dacon et al. These bind the fusion peptide.
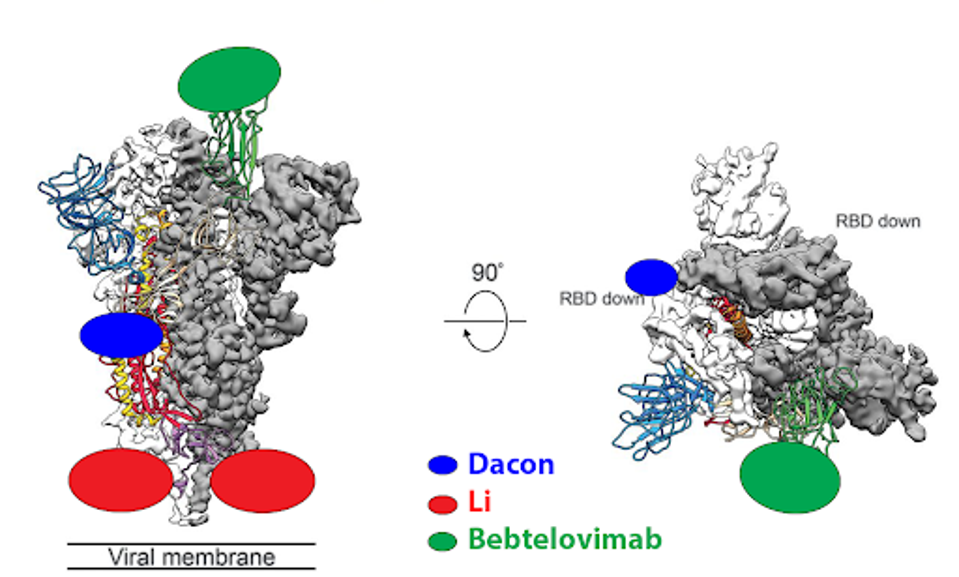
FIGURE 4: Binding epitopes of the bebtelovimab, Li, and Dacon antibodies.
ACCESS HEALTH INTERNATIONAL
Again, these antibodies are broadly neutralizing across many variants but fall short of binding affinities of a bebtelovimab or Evusheld. With the assistance of RESP, perhaps slight modifications to in-development antibodies could yield a powerful monoclonal antibody candidate primed to save thousands of lives.