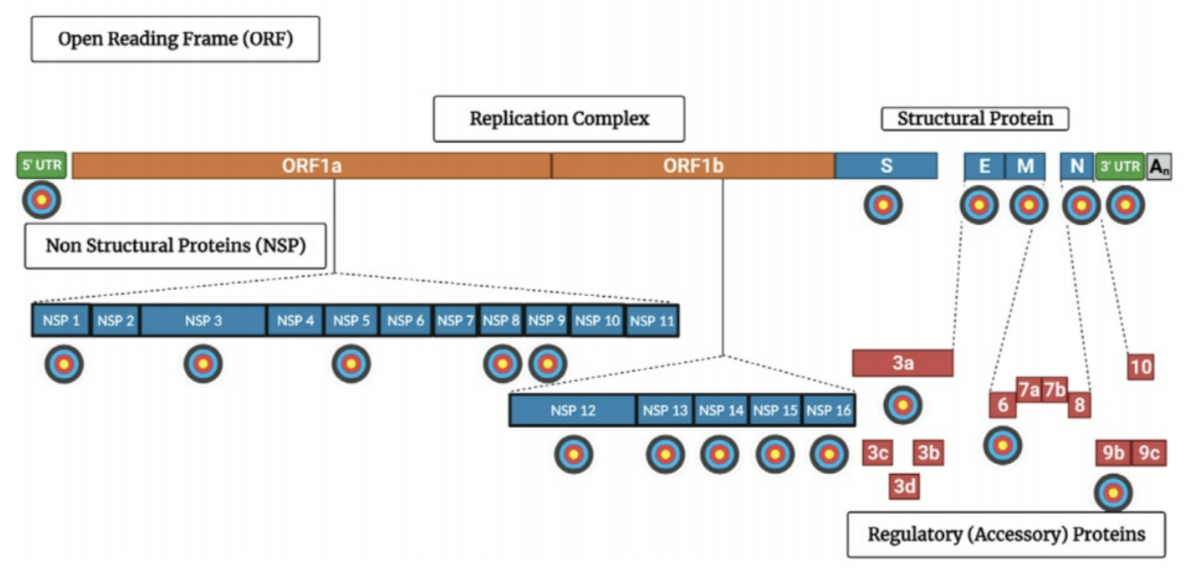
To survive and replicate inside our cells, a virus has to be able to adapt them to its own advantage. This means altering the cells’ metabolism, physiology, and gene expression. Often, these adaptations are part of a strategy to bypass and thwart our innate immune system — a virus can’t get very far if our immune system manages to recognize it early on. So, suppressing our immune response is one of a virus’ top priorities. SARS-CoV-2 is particularly crafty in this regard, possessing many different tactics that help keep it under the radar. But there’s a flipside to this craftiness, it is highly complex. A great deal of different processes and pieces need to come together for it to work. Block one, and the whole thing can come crashing down. This makes it an excellent avenue for therapeutic interventions. A group of researchers based at the Indian Institute of Science (IISc) have uncovered a new target: SARS-CoV-2 nonstructural protein 1 (NSP1). Published in eLife, their work shows that binding to and blocking NSP1 leads to a decrease in viral replication in human immune cells.
There are two major categories of viral proteins: structural and nonstructural. Structural proteins are the components of the virus particle itself. They provide the essential elements the virus needs to transmit from one person to another. Nonstructural proteins, on the other hand, are not a component of the virion particle itself. Instead, they are produced by the virus once inside host cells. Here, the nonstructural proteins help with viral replication by regulating transcription and impairing host defenses.
NSP1 is an especially versatile nonstructural protein, with a variety of different functions related to both immunosuppression and viral replication. It is one of the first viral proteins released once inside the cell. The primary function of NSP1 is to inhibit the translation of host messenger RNA (mRNA). To replicate, viruses need to suppress the genes of the host cell and instead favor synthesis of their own. NSP1 contributes to this by binding to ribosomes, which act as the “cellular machinery” via which proteins are made. More specifically, NSP1 is made up of two sections, an N terminal and a C terminal. It is the C terminal that binds deep into the mRNA entry tunnel of the small ribosomal subunit (40S). Once bound, NSP1 prevents host messenger mRNA from being translated, but in an act of delicate trickery, still allows for viral mRNA to be translated (Figures 1 & 2).
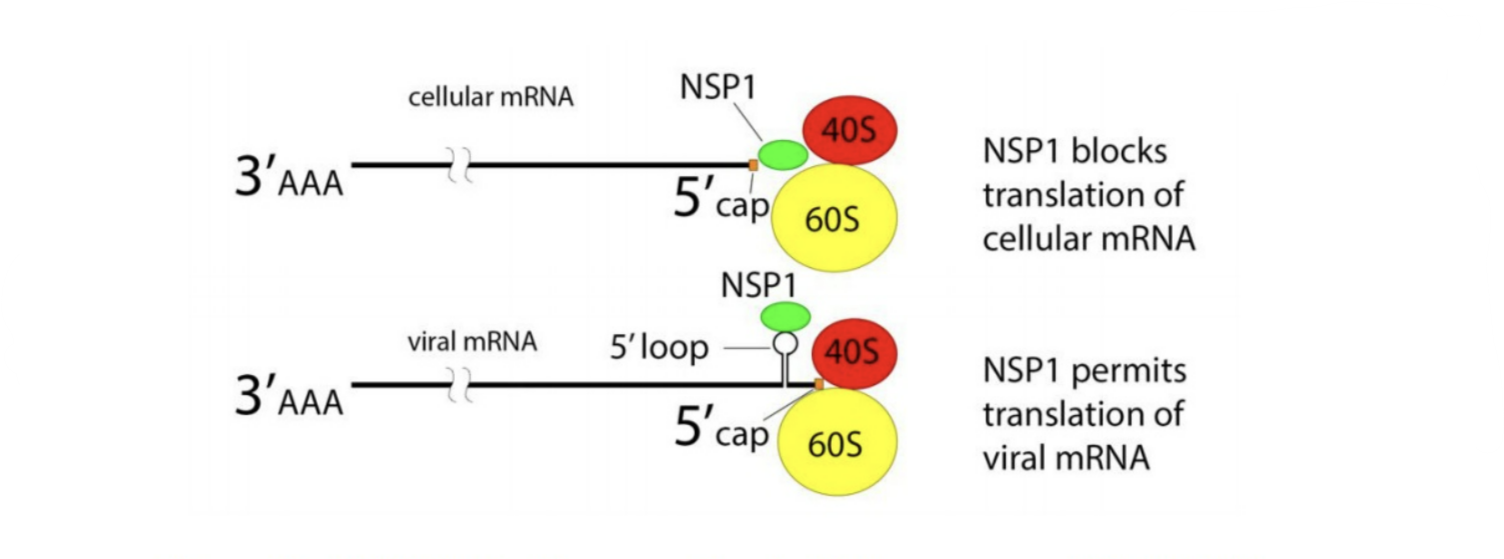

Downstream, the sabotaging of host mRNA translation means a variety of important antiviral proteins don’t get produced. This weakens the immune response our bodies can muster, and in a dangerous feedback loop, makes it even easier for the virus to replicate and spread unchallenged.
Aside from its pivotal role in viral replication and host immunosuppression, Asafar et al. focused on NSP1 because it is one of the SARS-CoV-2 proteins with the lowest mutational frequency. That is, it changes very little over time and across variants. Any drug that wishes to remain effective against SARS-CoV-2 has to contend with the rise of new variants — zeroing in on a highly conserved, functionally important protein like NSP1 improves the odds.
Once they’d chosen their intended target, the team of scientists combed through a database of around 1,600 FDA-approved drugs. Turning to computational modeling, they isolated drugs with the potential to bind the NSP1 C-terminal. Of a final list of a dozen or so candidates, Asfar et al. narrowed it down to one, montelukast.
Montelukast is usually used to treat asthma and hayfever. How might it work to curb SARS-CoV-2 infection? In a nutshell, by binding to the C terminal of NSP1 before NSP1 has a chance to bind to our ribosomes. This clogs up NSP1’s binding site and prevents it from inhibiting our cell’s mRNA translation.
At least, that’s how it should work in theory. To find out whether it worked in practice, the researchers engineered human kidney cells that express NSP1. They observed a noticeable drop in mRNA translation and protein synthesis. Next, Asafar et al. exposed these cells to montelukast. The drug restored protein synthesis to normal levels, successfully counteracting the effects of NSP1.
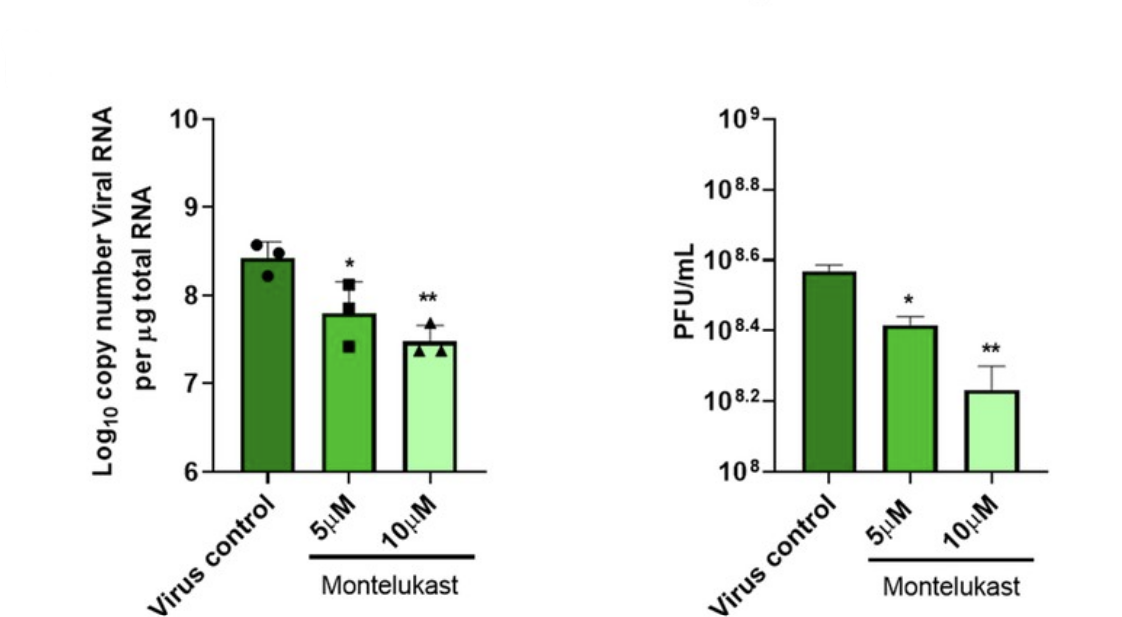
Asafar and his colleagues also exposed human cells to live SARS-CoV-2 virus and then treated those cells with montelukast. They witnessed a significant drop in the expression of SARS-CoV-2 spike protein, suggesting reduced viral replication. The results indicate that montelukast managed to prevent NSP1-mediated inhibition of host mRNA translation, allowing for normal production of antiviral proteins and a reduced capacity for viral replication (Figure 3).
Despite the positive results, the strength with which montelukast binds to NSP1 —known as “binding affinity”— is still relatively low. Asafar et al. admit that montelukast on its own likely won’t be up to the task. But its mechanism of action, the way in which it prevents SARS-CoV-2 NSP1 from inhibiting host mRNA translation, is a viable one, and a good starting point for future drug design. Importantly, it hints at an overall strategy for successful therapeutic intervention: isolating the specific processes by which SARS-CoV-2 manages to suppress and evade our immune response and discovering drugs that inhibit each one. The book, Natural Immunity and Covid-19: What it is and How it Can Save Your Life, provides a detailed overview of these processes. Figure 4 highlights the virus’ multiple points of sensitivity. I foresee a day when there will be twenty or more different classes of drugs (with multiple drugs in each class) each targeting a different viral protein. When used in combination, these drugs will be safe and effective in preventing and treating Covid-19, no matter what variant may arise.