This is the second in a series examining the past, present, and future of the pandemic and viral variants.
Each successive iteration of SARS-CoV-2 variant to date was more transmissible than the last. New variants drove multiple waves of Covid-19 throughout the world. Here, examine what happened in the past and present of the pandemic in order to understand what may lie in our future.
Having written the book Variants! The Shape-Shifting Challenge of Covid-19, one of the most frequent questions I am asked is ‘what comes next?’ To begin to answer this question, we examined the underlying dynamics of virus variation in part one. Here, in part two, we will delve more deeply into the topic with an analysis of the Gamma variant in Brazil.
A recent paper by Naveca et al. analyzes the virology of the Brazilian SARS-CoV-2 experience. The Amazonas region of Brazil experienced two major waves of infection and Naveca et al. used deep genomic sequencing to understand the strain of virus causing these waves.

Generating 1,188 SARS-CoV-2 whole-genome sequences from infected Amazonas residents from January to July, the researchers confirmed that 99.7% of infections were driven by the Gamma variant. However, the researchers found several mutations in their sequences uncommon to the traditional Gamma variant, which is illustrated in Figure 2.
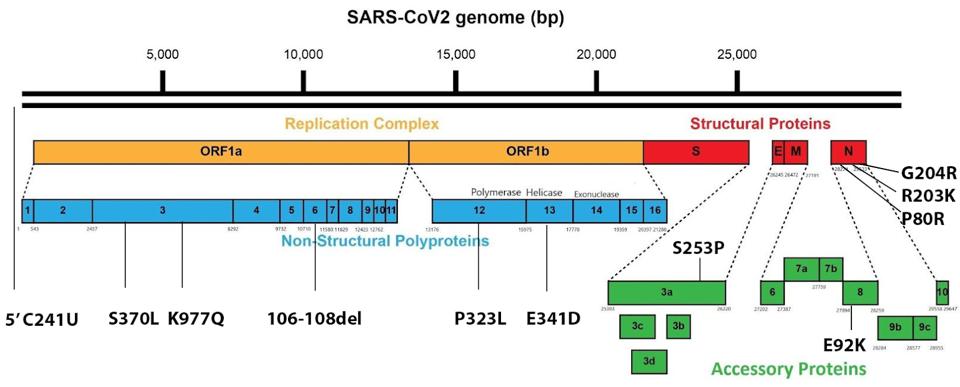

Although there was a major initial wave of the P.1 original Gamma variant, Naveca et al. describe what they believe are next-generation variants, which they collectively refer to as “Gamma Plus.” The researchers hypothesized that the endemic state of the original Gamma variant in Amazonas allowed the rise of these second-generation variants. The Gamma sublineages contain mutations that enabled not only increases in transmissibility, but the potential for vaccine breakthrough cases. These factors strongly indicated that the Gamma sublineages drove the Gamma wave of infections in Brazil further than the solely the original Gamma variant. As many as seven “Gamma Plus” variants were detected that carried all the mutations of its parent, but a few more in key regions of the virus. Even though Gamma infections are waning currently in Brazil, it is worth analyzing the Gamma Plus variants to see how a dominant variant can vary still further to drive higher levels of infection and death.
Naveca et al. summarize that continuous evolution of the P.1 (Gamma) variant of concern was the likely cause of steady-state SARS-CoV-2 cases from May to July 2021, and were partially responsible for infections as early as January. This period resulted in the most Covid-19 related deaths, both regionally in the Amazonas region and throughout the whole of the country.


What are the Gamma Plus sublineage mutations that Naveca et al. associate with still higher levels of infection than Gamma? The researchers’ attention was primarily directed to the Spike protein, in two regions in particular: the N-terminal domain and the Furin cleavage region. In the N-terminal domain, the researchers note deletions at positions 144, 142-144, and 141-144. These mutations are observed in major variants of concern or interest such as Alpha and Eta, as well as other circulating variants like C.1.2 and A.30. The researchers estimate that roughly 10% of infections by Gamma sublineages contain one iteration of these N-terminal domain mutations, most of which are conveyed in the P.1.3 variant.

In addition to playing an important, but a nonspecific role in attracting the virus to the host cell surface, the N-terminal domain is critically involved in the antigenic binding of a large portion of neutralizing antibodies to the Spike protein.
This fits with the history of many variants. Among those that undergo deletions or insertion of positive charges via point mutations in this region to avoid neutralizing binding by antibodies and convalescent sera are Alpha, Beta, Delta, and Lambda. The deletions in the Gamma sublineages may work to accomplish a similar feat.

The second of the major modified regions in Gamma sublineages is the Furin cleavage site.

Most coronaviruses have a single cleavage site called “S2.” This site is activated when the virus binds to its target. Some coronaviruses, such as SARS-CoV-2, have an additional cleavage site known as the “Furin cleavage site.” Furin is a protease enzyme which cleaves the SARS-CoV-2 virus as it buds from the cell. This cleaving is what activates the Spike protein. The Spike protein must be activated to initiate viral entry. In the original Wuhan strain of SARS-CoV-2, cleavage is not 100% efficient, meaning virus particles are not uniformly cleaved. Mutations in this region can improve this efficiency, resulting in more consistent cleavage of SARS-CoV-2 particles, and yielding a more infectious virus.
Near the Furin cleavage site, the researchers found two mutations in four of the Gamma sublineages. The first Gamma sublineage mutation in this region is N679K. Notably unobserved in major variants of concern or interest, this mutation is found in the C.1.2 variant originating in South Africa. Asparagine to lysine is a major change in charge, from neutral to positive. This means the Furin cleavage region in P.1.3 and P.1.4 is changed significantly both in charge and structure, potentially reinforcing cleavage.
The second mutation that the researchers note is P681H/R. This mutation has been previously noted in Alpha, Mu, and A.30. Additional mutations at this position are noted in Delta, Kappa, and A.23.1, showing that modifications to this position likely result in enhanced Furin structure. Proline to histidine is another neutral to positive shift, also pointing to a net positive increase in regional charge. The figure below demonstrates the positive charge shift due to the Furin cleavage mutations.

Again, this follows a trend of previous variants containing Furin cleavage region mutations, such as Alpha, Delta, and Kappa, which demonstrate mutations at position 681. We can expect new variants to continue these trends. Both the N-terminal domain and the Furin cleavage region are very flexible, via positive charge alteration or antigenic site deletions, the virus will change whatever it needs to enhance its capabilities. We should monitor mutations in these regions for both neutralization and transmission in tissue culture.

We note that the mutations in th Gamma Plus variants are found in the Furin cleavage region and N-terminal domain, but not the heavily examined receptor-binding domain. There is one minor amino acid cange in P.1.8, but this is among the least prevalant Gamma Plus sublineages. The major lineages, P.1.3 through P.1.7, all lack receptor-binding mutations.
In addition, we note that while the Spike protein is certainly important, there are other mutations in the nonstructural proteins and regulatory Orfs that must be considered.
In the Orf1ab replicative proteins, the researchers observed six mutations in four of the seven sublineages analyzed. In the NSP3, there are three observed mutations. The first is T133I, found in the Acidic C-terminal domain. The second is I441V, found in the SARS-specific unique domain. The third is T1018I, found in the papain-like protease, which is the most important of the NSP3 subdomains and oversees translational cleavage of NSP1-4.
The researchers noted A446V in NSP4 of one of the sublineages. NSP4 is vital to the formation of the double-membrane replication vesicle. The final Orf1a mutation is T141M in NSP8, which provides stability to the polymerase replication complex.
In NSP12, the RNA-dependent RNA polymerase of SARS-CoV-2, the researchers note P918L. This protein is the engine of the transcription-replication complex, reading nucleotide sequences and copying them for the creation of new RNA. We note that all mutations in the Orf1ab replicative proteins are unique to the Gamma Plus variants among major variants of concern or interest to this point.
There are no mutations to the structural proteins of the sublineages, but there are four noted mutations in the regulatory (accessory) proteins that play a role in viral pathogenesis. In Orf3a, the protein that oversees apoptosis, or cell death, the researchers note the S261L mutation. In Orf7b, which is involved in the subversion of interferon signals like STAT2, as well as modulation of virus particle interaction with the cell via tetherin interaction, the researchers note the M24T mutation. Finally, in Orf8, which downregulates the major histocompatibility complex (MHC-I), the researchers note H17Y and G8stop. The G8stop mutation is notable in the P.1.8 subvariant as this effectively destroys the whole of the Orf8 protein, rendering its functions incapacitated. The regulatory protein mutations described here are unique to the Gamma Plus variants, as we noted about the replicative protein mutations.

Variants are continuously selected from increased transmissibility and because Delta is now overtaking Gamma and its sublineages, that suggests that Delta is a far more transmissible virus than variants that came before. We can use our understanding of Gamma and its sublineages to further examine the more transmissible Delta, its own sublineages, and their higher transmissibility and immune evasion.