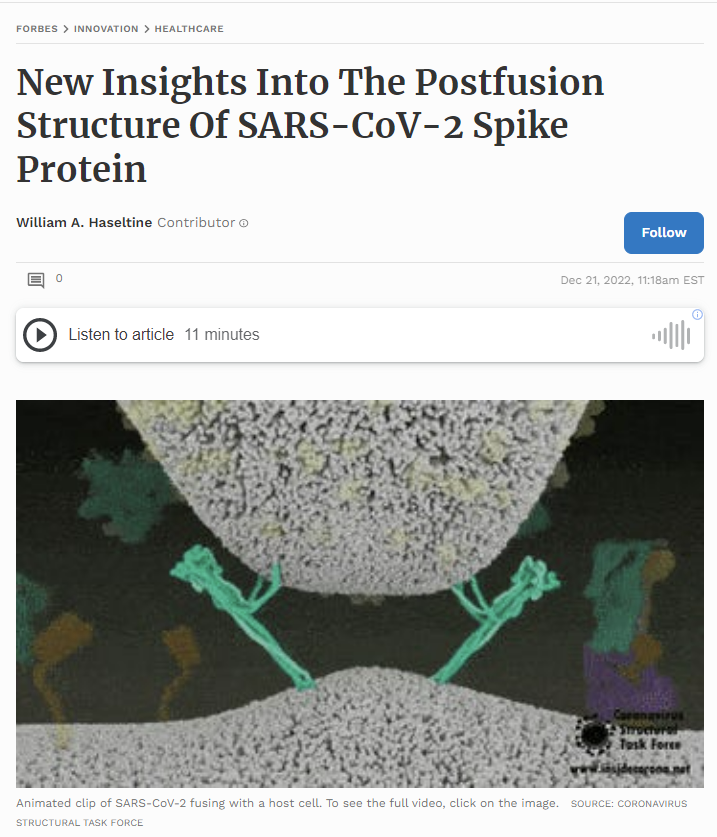
Covid begins when the SARS-CoV-2 virus first encounters cells in the upper airway that express a suitable attachment site, the angiotensin converting enzyme 2 (ACE 2). Blocking binding and entry short circuits infection before it begins. That is why vaccines can be so effective in protecting us from disease. However, as we are learning, the duration of vaccine protection is so short and specific to each virus variant that we cannot quell the pandemic with vaccines alone. We need new highly potent, long-acting, safe antiviral drugs to protect us and end the pandemic. In my opinion the only way to find these drugs is to understand the virus in all its complexity. Fortunately, we have the tools of modern biomedicine to do so if we wish. A recent preprint from the Department of Molecular and Cellular Biology at Harvard University provides atomic-level information revealing new and unexpected aspects of the early critical steps of viral entry.
Opening the Gates: Membrane Fusion
Entry into the target cells requires much more than surface attachment. Both the virus and host cell are surrounded by a protective membrane.
Viral entry begins when the virus binds to the surface of the target cell via the ACE 2 protein on the target cell surface. Binding is followed by fusion of the virus cell membrane to that of the host cell. Both binding and fusion are mediated by the tripartite spike protein embed in the virus membrane.
The spike protein is originally made of a single long polypeptide. As the virus exits the cell the spike protein is usually cut at what is called the furin cleavage site, producing the amino-terminal S1 protein and the carboxyl-terminal S2 protein. The actual spike is a timer consisting of three S1 and three S2 proteins. In the pre-fusion state, the S1 protein wraps around, covering almost the entirety of the S2 protein. In Figure 1, regions of the spike protein gene are highlighted in different colors and identified by abbreviations that denote their function. Figure 2 depicts the structure of the pre-binding S1/S2 trimer.
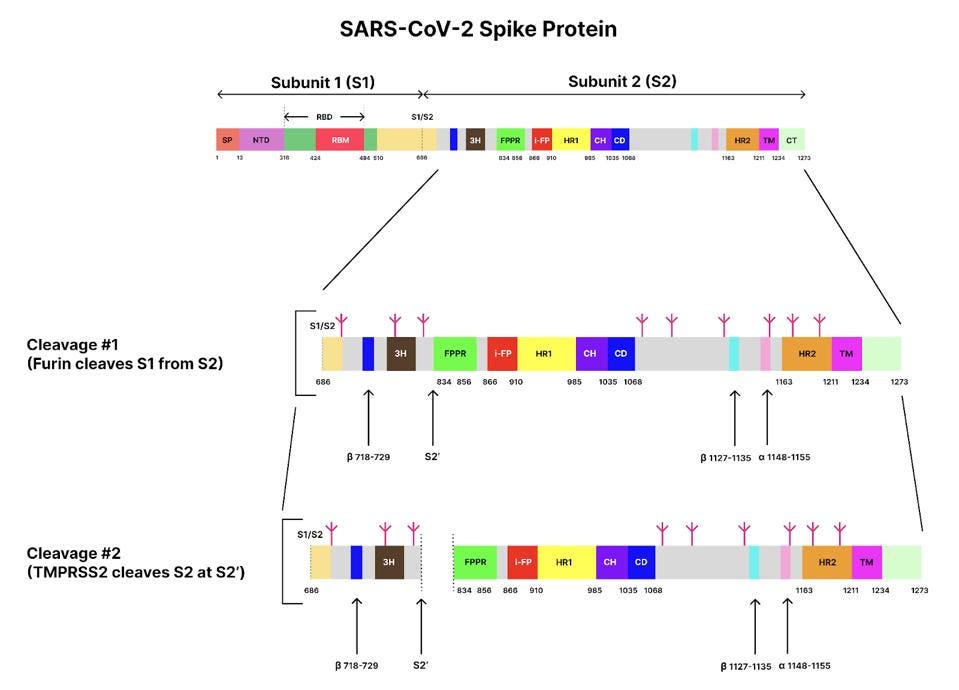
FIGURE 1. Schematic of the SARS-CoV-2 spike protein genome with its different domains and segments. S1/S2 represents the furin cleavage site, and S2’ represents the transmembrane serine protease 2 (TMPRSS2) cleavage site. Glycans are indicated by small, magenta tree-like symbols. Subunit 1: SP, signal peptide; NTD, N-terminal domain; RBD, receptor-binding domain. Subunit 2: 3H, three-helix segment; FPPR, fusion peptide proximal region; i-FP, the internal fusion peptide; HR1, heptad repeat 1; CH, central helix region; CD, connector domain; β1127-1135, a β-strand formed by residues 1127-1135; a1148-1155, an a-helix formed by residues 1148-1155; HR2, heptad repeat 2; TM, transmembrane anchor; and CT, cytoplasmic tail. SOURCE: ACCESS Health International (Adapted from: “Cryo-EM structure of SARS-CoV-2 postfusion spike in membrane” SHI ET AL. 2022)
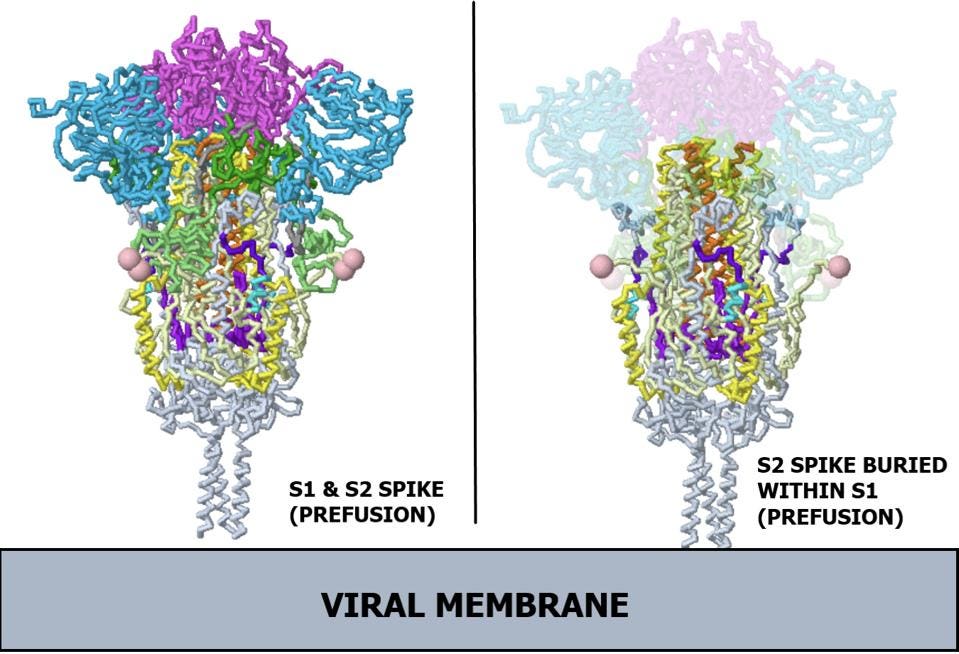
FIGURE 2. Ribbon diagram of SARS-CoV-2 spike protein. (Left) Complete prefusion spike protein, including both subunits. (Right) X-ray view of prefusion S2, buried within S1. Note, the receptor binding domains are at the top. The bottom extends through parts missing in this model: stem, transmembrane domain, cytoplasmic domain. Pink balls mark the furin cleavage sites that separate S1 from S2. S1 is translucent in the image at right. S1 domain key: Receptor binding domain (RBD) in magenta; N-terminal domain (NTD) in light blue; C terminal region of S1 fragment after furin cleavage (CTD1) in dark green; C terminus of S1 fragment after furin cleavage (CTD2) in light green. S2 domain key: Fusion peptide (FP) in teal; Fusion peptide proximal region (FPPR) in dark purple; Heptad repeat 1 (HR1) in yellow; Central helix (CH) in orange; Connector domain (CD) in light purple. SOURCE: ACCESS Health International (Adapted from Proteopedia, based on Cryo-EM work from “Distinct conformational states of SARS-CoV-2 spike protein” CAI ET AL. 2020)
Upon binding to the ACE2 receptor on the target cell surface, all three S1 proteins separate from the complex, releasing the S2 trimer. The S2 protein is then cut by a cellular enzyme into a short amino-terminal fragment, which here we will call the SAT fragment, and the longer S2 protein. It is the S2 protein-SAT fragment complex that drives the viral-cell membrane fusion. Figure 3 illustrates the structure of one of the three S2-SAT fragments of the pre-fusion protein free of the surrounding S2. Note that the SAT-fragment remains tightly associated with the S2 protein post the second cleavage.
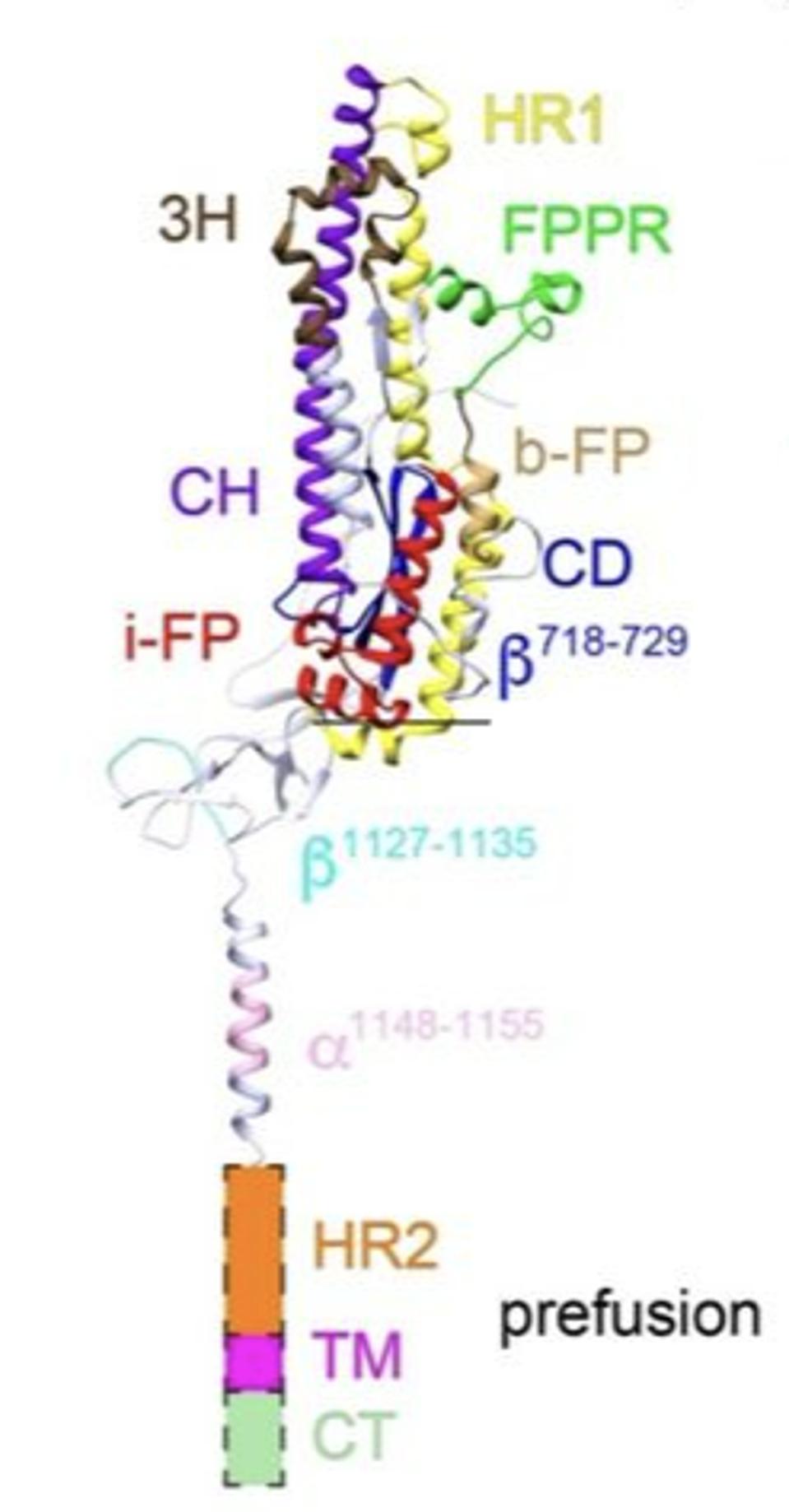
FIGURE 3. Prefusion structure of one of three subunit 2 protomers of the SARS-CoV-2 spike protein, after subunit 1 has been removed by ACE2. Labels: 3H, three-helix segment; FPPR, fusion peptide proximal region; i-FP, the internal fusion peptide; b-FP, “bona fide” fusion peptide; HR1, heptad repeat 1; CH, central helix region; CD, connector domain; β1127-1135, a β-strand formed by residues 1127-1135; a1148-1155, an a-helix formed by residues 1148-1155; HR2, heptad repeat 2; TM, transmembrane anchor; and CT, cytoplasmic tail. FROM: “Cryo-EM structure of SARS-CoV-2 postfusion spike in membrane” SHI ET AL. 2022
Fusion of two membranes, once begun, is an energetically favorable reaction. However the charged surfaces of the membranes repel one another and require additional energy to initiate the fusion process. That energy takes several forms. The initial step is provided by the S2 protein. Once free of S1, it springs into a fully open position, like a jackknife , embedding its amino-terminus into the juxtaposed host cell membrane, effectively harpooning the cell and tethering the virus to the cell. Some additional energy for the eventual fusion is derived from the association of the amino-terminus of the S2 protein as it buries itself in the cell membrane. The actual structure of this open structure remains to be resolved.
We know much more about the next step. The S2-SAT fragment complex then folds back on itself, pulling the two membranes together (Figure 4). The energy for this reaction comes from the formation of a very stable final structure whereby each S2 complex creates a long alpha helix folded back upon itself. The final structure is a six-helix bundle embedded in both the viral and cell membranes. Multiple spike proteins working side-by-side in parallel create an open pore that then widens to permit entry of the virus genetic material into the interior of the cell.
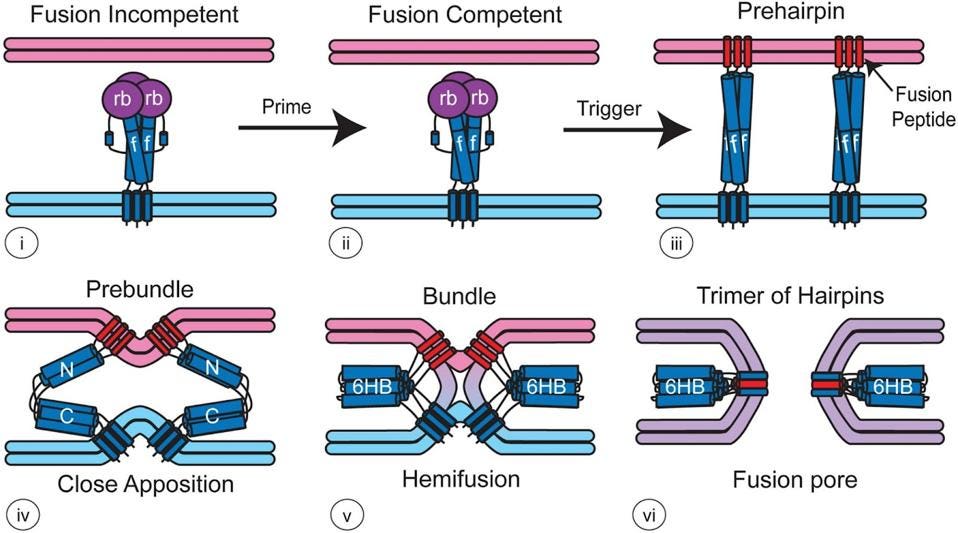
FIGURE 4. Model for how viral fusion proteins function — For most class I fusion proteins, prior to triggering (i and ii), the receptor-binding subunit (deep purple, rb) clamps the fusion subunit (dark blue, f). Upon triggering, the receptor-binding subunit moves out of the way unclamping the fusion subunit so that it can form a prehairpin embedded in the target membrane via the fusion peptide (red). The prehairpin then folds back causing the N- and C-α-helical heptad repeats to form a six-helix bundle (6HB) and progressively pulling the target (pink) and viral (light blue) membranes through stages of close apposition (iv), hemifusion (v) and fusion pore formation (vi) FROM: “Fusion of Enveloped Viruses in Endosomes” WHITE & WHITTAKER 2016
Postfusion Structure of the SARS-CoV-2 Spike Protein
Although we have a solid sense of what the prefusion spike protein looks like, until now the same could not be said about the postfusion spike protein. Given the importance of membrane fusion to the viral life cycle, this represented a serious lacuna.
Enter cryogenic electron microscopy (Cryo-EM), a technique that helps researchers determine the structure of biological molecules. In a nutshell, a sample of the target molecule is flash-frozen in a solution. The frozen solution is then blasted with an electron beam, which hits the molecules and passes through a downstream lens that creates a magnified image of the structure on a detector plate. A camera takes thousands of two-dimensional images which, with the help of algorithms, can be layered into a three-dimensional model. The benefit of cryo-EM is that researchers can easily flash freeze the same protein at various different conformational stages, giving them a sense of how the structure changes over time.
Shi et al. prepared the SARS-CoV-2 spike proteins for cryo-EM by wrapping them up in a lipid bilayer using scaffolding proteins, creating a so-called “nanodisc” — the scaffolding protein keeps the lipid bilayer, which mimics a cell’s membrane, tightly pressed up against the spike protein (Figure 5). Next, they exposed the nanodisc spike constructs to ACE2, triggering conformational changes and membrane fusion. After the first cleavage event, subunit 1 falls away while still attached to ACE2, leaving S2 exposed.

FIGURE 5. Schematic diagram of a membrane scaffold protein nanodisc. The membrane scaffold protein is highlighted in green, the lipid bilayer in gray, and the target membrane protein in orange. SOURCE: Cube Biotech
The researchers placed the dissociated, nanodisc-wrapped S2 under the electron microscope. The resulting structure can be seen in figure 6, below.
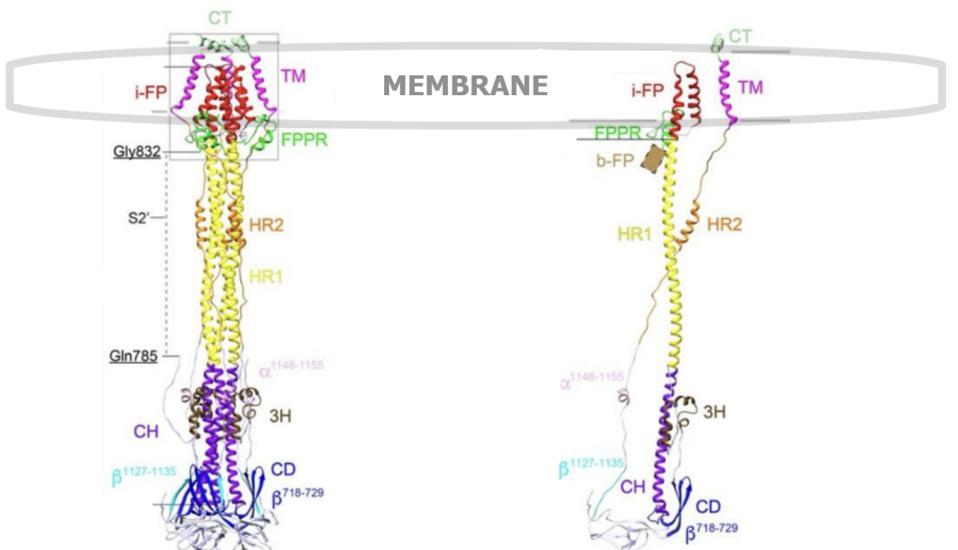
FIGURE 6. (Left) A ribbon diagram of the SARS-CoV-2 S2 spike protein trimer, postfusion. (Right) Same S2 spike protein postfusion, but one single protomer in isolation. Labels: 3H, three-helix segment; FPPR, fusion peptide proximal region; i-FP, the internal fusion peptide; b-FP, “bona fide” fusion peptide; HR1, heptad repeat 1; CH, central helix region; CD, connector domain; β1127-1135, a β-strand formed by residues 1127-1135; a1148-1155, an a-helix formed by residues 1148-1155; HR2, heptad repeat 2; TM, transmembrane anchor; and CT, cytoplasmic tail.SOURCE: ACCESS Health International (Adapted from: “Cryo-EM structure of SARS-CoV-2 postfusion spike in membrane” SHI ET AL. 2022).
Homing In on the Fusion Peptide
In an exciting “first”, Shi et al. successfully managed to resolve the membrane-interacting sections of the S2 fragment.
Their work settles a long-standing point of debate regarding the final post fusion structure. Three different regions had previously been suggested as potential candidates for the fusion peptide, but none had been acceptably confirmed. The first is located upstream of the S2’ cleavage site, called the n-terminal fusion peptide (n-FP). The second candidate, just downstream of the S2’ cleavage site, has been called the “bona fide” fusion peptide (b-FP) — this area is highly conserved across coronaviruses. And finally, a region just upstream of heptad repeat 1 (HR1), called the internal fusion peptide (i-FP).
Although the so-called “bona fide” region had previously been considered the likeliest candidate, Shi et al. show that it is actually only the internal fusion peptide (i-FP) that fully enters the target lipid membrane; neither of the other two candidates insert into the cell membrane in the postfusion conformation, making it unlikely that either acts as the true fusion peptide.
The i-FP region of the postfusion structure extends from the central S2’ coil, creating one long, rigid alpha helix. Once inside the lipid bilayer, the internal fusion peptide makes a sharp U-turn back towards the outside of the cell membrane, creating a hook or hairpin shape. When the three protomers are put together in their natural trimeric conformation, the fusion peptides of the S2 come together to form a cone-like structure — the researchers speculate that this cone shape helps the spike protein penetrate the target cell membrane, like the tip of an arrow.
Fusion Peptide as Docking Site
Along with settling the location of the fusion peptide, Shi et al. also managed to solve another enduring question: does the fusion peptide interact with the transmembrane segment during fusion? It had been speculated that the two may link during the final stages of the fusion process, but again, direct structural evidence had been missing.
Indeed, their work shows that the cone formed by the internal fusion peptides is bound by three transmembrane segments, forming a larger, nine-helix-bundle cone. This larger, transmembrane cone is itself capped by three copies of the cytoplasmic tail segment, which lie flat across the top (Figure 7). Both the transmembrane region and the cytoplasmic segment further stabilize the postfusion construct.
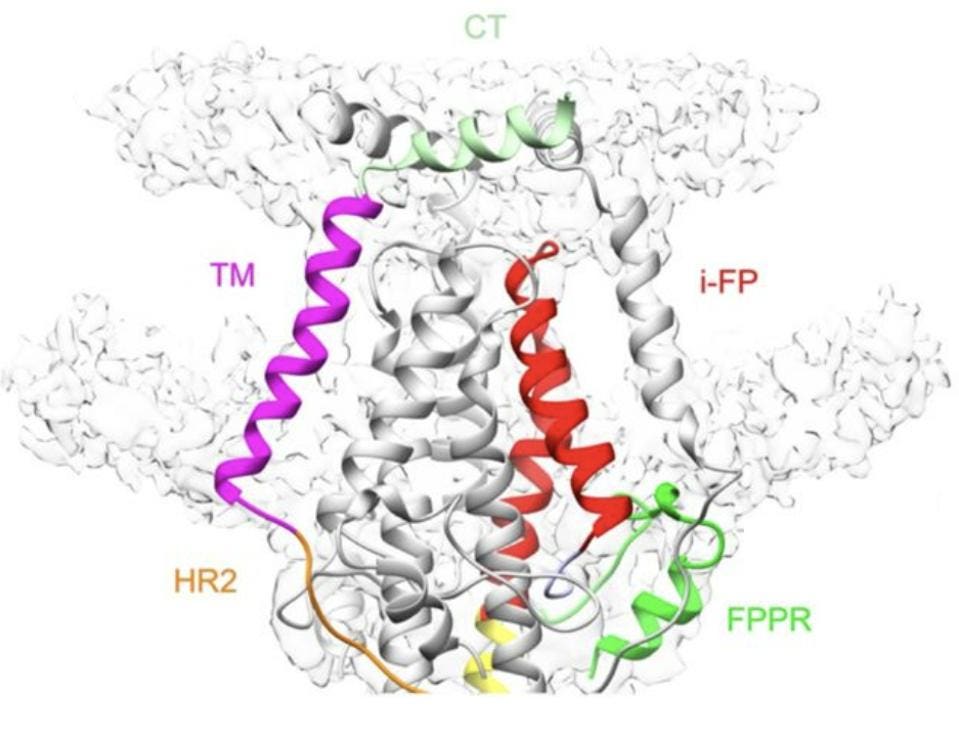
FIGURE 7. Ribbon diagram of the transmembrane region of the postfusion S2, with the cytoplasmic tail clearly visible, capping the structure. Heptad repeat 1 (HR1) in yellow, fusion peptide proximal region (FPPR) in green, internal fusion peptide (i-FP) in red, Heptad repeat 2 (HR2) in orange, transmembrane segment (TM) in magenta, and cytoplasmic tail (CT) in light green. FROM: SHI ET AL. 2022
An area previously considered separate to the transmembrane segment, called the pre-transmembrane segment, is shown in this model to lie fully within the membrane, extending the anchor by 13 amino acids and forming one single transmembrane structure.
Implications
In what can only be described as a tour de force, Shi et al. have given us a clearer picture of the SARS-CoV-2 spike protein in its postfusion conformation. Not only is this a first in the study of coronaviruses, but also in the study of class I fusion proteins as a whole. Using cryogenic electron microscopy, the researchers solved two enduring mysteries: the exact location of the fusion peptide, and whether or not the fusion peptide and the transmembrane segment interact.
An unanswered question and a caveat remain.
The first step of the fusion process is harpooning of the target cell by the S2 protein. The structure of the initial fusion intermediate is not resolved. We are free to speculate what it might be.
A caveat is that the structural model developed by Shi et al. is solved for the S2 protein absent the S2’ cleavage (see figure 1, first and second cleavage). That leaves 147 additional amino acids covalently attached to the S2 protein. These are located immediately amino-terminal to what was previously known as the “bona fide” fusion peptide. Might the result of either the first intermediate or the final postfusion structure differ were amino acids 834-856 terminus of the S2 in the Shi et al. experiments to more closely resemble that of what is almost certainly the actual S2 protein of the infectious virus?