This is the first of a two-part series. Here we describe the mutations in the Omicron Spike protein. In part two, we will describe mutations in other parts of the genome.
A new variant, Omicron, very effectively evades the neutralizing antibodies of the Pfizer vaccine regimen, according to a recent study. The research by Cele et al. demonstrates that neutralization protection dropped over 40-fold against the Omicron variant as compared to the D614G Triad strain. This includes those with the highest protection, having been previously infected and then two-dose vaccinated with the Pfizer vaccine. In other words, a two-dose regimen of the Pfizer vaccine will not protect against Omicron. Whether three doses is protective, we do yet not know.

The Omicron variant has the potential to spark the next great wave of SARS-CoV-2. Dozens of countries are on high alert as Omicron sequence detection is rapidly increasing. Case numbers are expected to rise dramatically in the coming days and weeks. See the rise of the Omicron variant in South Africa and globally below.

How does Omicron evade vaccine neutralization? The variant carries at least 50 amino acid-altering mutations and at least 10 silent mutations in coding and non-coding regions. At least 36 of these mutations are found in the Spike (S) protein, which is a major modulator of infectivity and immune evasion. Many of these mutations are shared with other major variants of concern or interest and have been previously shown to increase infectivity and decrease recognition by vaccine-administered and monoclonal antibodies. Other mutations are unique to this variant. All of which may contribute to the increased functionality of the virus.
Early returns on transmissibility, including the rate of virus replication, suggest it is high. Omicron is outpacing many earlier variants in terms of new infections, such as the Beta and Delta variants described below.

Omicron additionally shares several amino acid mutations with other major variants, described in the figure below. Here we are going to focus on the S protein. In the second part of this two-part series, we will discuss the interaction of non-S proteins and their contributions to Omicron’s enhanced virological characteristics.

Spike Protein (S)
The SARS-CoV-2 S protein binds the host ACE2 receptor and initiates infection. The Omicron variant S protein is heavily mutated as compared to other major variants. It contains a staggering 36 amino acid-altering mutations, 23 of which have been previously observed in other variants. We call this the sum of all fears because Omicron contains almost all of the S protein mutations that resulted in previous variants of concern, plus a large compliment of its own.
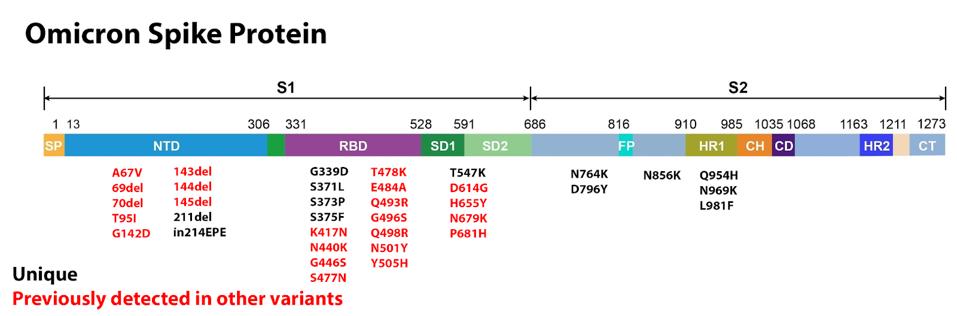

The first major domain in the S protein is the N-terminal domain. This domain is a target for both binding and neutralizing antibodies in convalescent sera and monoclonal treatments. Deletions are often found in the N-terminal domain, usually indicating the destruction of a binding site, meaning the variant becomes more resistant to antibody binding. There are six deletions in the Omicron variant: 69, 70, 143, 144, 145, and 211. The most likely explanation for these deletions is immune evasion. The only deletion unique to Omicron is 211, whereas all others are found in other variants such as Alpha, Gamma, or Delta.
There are two potential explanations for the same mutations occurring in multiple strains. One is convergent evolution, in which strains develop the same mutation independent of each other, and the second is recombination, where two strains exchange genetic material during some cross-infection event. In other words, it is possible Omicron picked up the deletions from another variant during its evolution.
Other mutations in the Omicron N-terminal domain include A67V, which was previously observed in the Eta variant originating in Nigeria, T95I, previously observed in the Iota variant originating in New York, and G142D, observed in the pervasive Delta variant. Because these mutations appeared in regional variants all over the world independently, we hypothesize they have some significant effect on the binding capability of antibodies, meaning Omicron has the potential to be highly resistant to antibody treatment.
Additionally, there is an insertion at position 214 which adds three amino acids to the genome. Some have speculated that this insertion is related to recombination with human cold-causing coronaviruses, specifically HCoV-229E. We offer a more likely alternative, suggested by Dr. Roberto Patarca, which is that the change is potentially due to internal recombination and translocation of a segment in the (-) strand of the 5’-untranslated region (UTR) of SARS-CoV-2 that would also explain other insertions at the same position 214. We note that this is a frequent site for insertions in other viruses.

The receptor-binding domain is the other major domain of note in the S protein. This is the region that makes direct contact with the host ACE2 receptor and plays a major role in transmissibility. The Omicron receptor-binding domain contains 15 amino acid substitutions, 11 of which have been previously noted in other variants.
Among the most concerning receptor-binding domain mutations found in Omicron are E484A, N501Y, S477N, and K417N. All of these have been previously observed in major variants of concern like Alpha and Delta. More interesting, however, is the overlap between some Omicron mutations and mutations found in wastewater variants in New York City. The collective mutations are noted to increase the overall positive charge of the receptor-binding domain, yielding salt bridges that boost ACE2 binding affinity.
In our previous article on Omicron’s possible origins, we noted the potential of cross-species infection of SARS-CoV-2 resulting in new and aggressive mutations. One study by Johnson et al. revealed dozens of mutations found in wastewater samples likely originating from rats or dogs. Several mutations found in those samples are in positions mutated in Omicron, including K417N, N440K, S477N, E484A, G496S, Q498R, N501Y, and others. This mass overlap may indicate that Omicron may have formed some or all of its mutations in a non-human animal before reinfecting human populations in Africa.
With respect to mutations falling outside the receptor-binding and N-terminal domains, there are those that fall near the furin cleavage site. Omicron shares a number of furin cleavage region mutations with other variants of concern and interest. T547K, H655Y, N679K, and P681H all play some role in boosting the efficiency of furin cleavage, which is a function of some viruses to cleave the S protein into two subunits, boosting transmissibility. These mutations have been previously observed in the Alpha, Gamma, Delta, or wastewater variants.
A number of mutations occur in the S2 subunit. These are N764K, D796Y, N856K, Q954H, N969K, and L981F. This region is critical for pre-and-post-fusion structures. It is likely that these mutations could contribute to increased infectivity by facilitating S protein fusion.
Lessons
One lesson to learn from Omicron is that there is a much wider range of variations for naturally-occurring strains in human populations. Omicron is very unlikely to be the last heavily mutated variant of concern. We should be prepared for that eventuality. Another lesson is that we need to attempt much more research on the origins of variants. Finally, we need to carefully characterize more than just individual proteins. In the second part of our Omicron discussion, we will examine the rest of the viral proteins and how mutations outside the S protein are just as important to analyze in reference to their impact on virulence and pathogenesis.