
The SARS-CoV-2 Omicron family of variants differs from the original isolates as well as other variants. Omicron viruses contain a minimum of 30 mutations in the Spike protein and another 23 elsewhere in the genome. Figure 1 illustrates how significantly mutated one of the currently circulating Omicron strains, BA.2.12.1, compares to the wildtype Wuhan virus. There are also silent mutations that change the nucleic acid sequence without changing the protein-coding capacity.
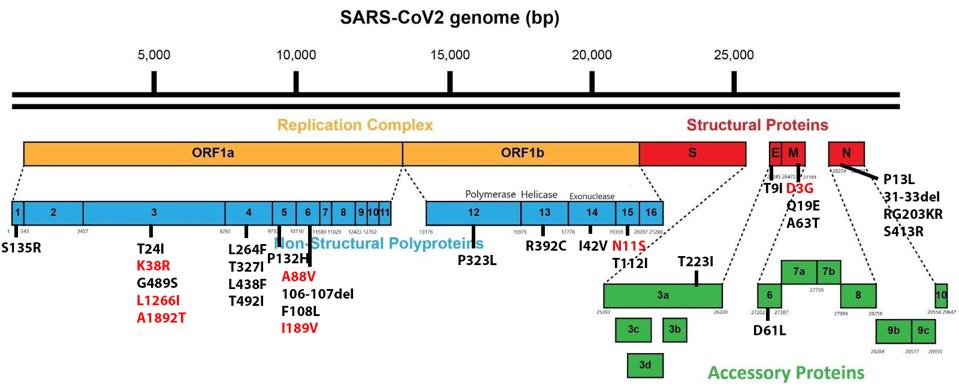
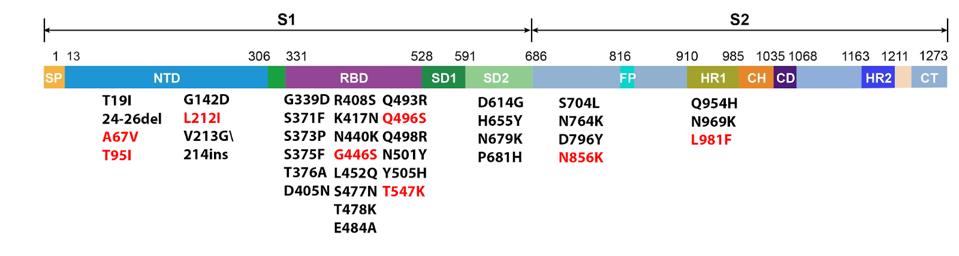
In addition to looking different in terms of amino acids, Omicron variants behave differently as well. Omicron is by far the most transmissible variant, including both rate of infection globally and infectivity between individual hosts. The family is diverse and at least three of the variants are at epidemic proportions: BA.4 and BA.5 in South Africa, as well as BA.2.12.1 in the US. While the disease spectrum still requires further examination, it is clear that Omicron can cause severe disease at high rates in children and the elderly, even in fully vaccinated individuals.
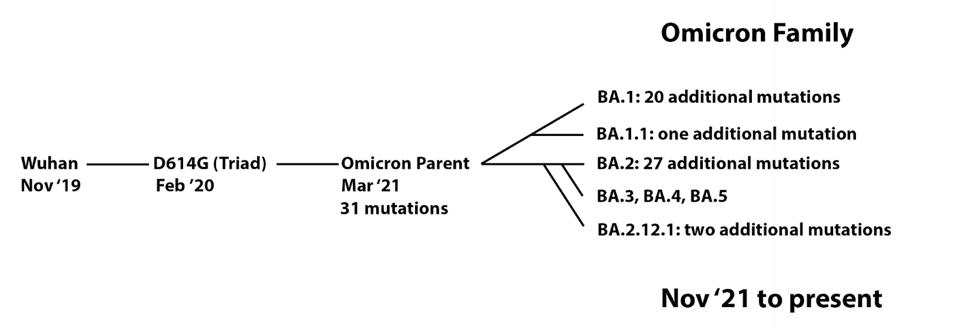
One of Omicron’s most salient features is how resistant the variant family is to vaccines and most monoclonal antibodies. Resistance contributes, at least in part, to the rapid spread of the Omicron variants. Previously, we described how amino acid changes in the Spike protein alter immune recognition by patient sera and monoclonal antibodies. Those infected with BA.1 can be reinfected with BA.2 and those infected with BA.2 can be reinfected by BA.4, BA.5, or BA.2.12.1.
More Tightly Packed Omicron Spike Protein
Wang et al. detail how multiple amino acid changes in the Omicron Spike protein not only eliminate antibody binding sites but also alter shape and function The Omicron Spike protein is significantly more compact than that of the original Wuhan (D614G) variant and the full range of variants that followed. Figure 3 compares the structure of the Omicron Spike with that of the Wuhan (D614G) wildtype. The Omicron protein is illustrated in green overlaid with the D614G variant in grey. The view is from the apex of the Spike looking toward the base. Major structural rearrangements are evident throughout the structure. I speculate that some antibodies which bind the looser structures fail to bind the Omicron Spike not only because of single amino acid changes but also as a consequence of the macro differences in structure. I also speculate that tighter packing implies that it has a more entropic structure, representing a higher energy state. That additional energy may be released during membrane fusion, increasing the efficiency of viral entry.
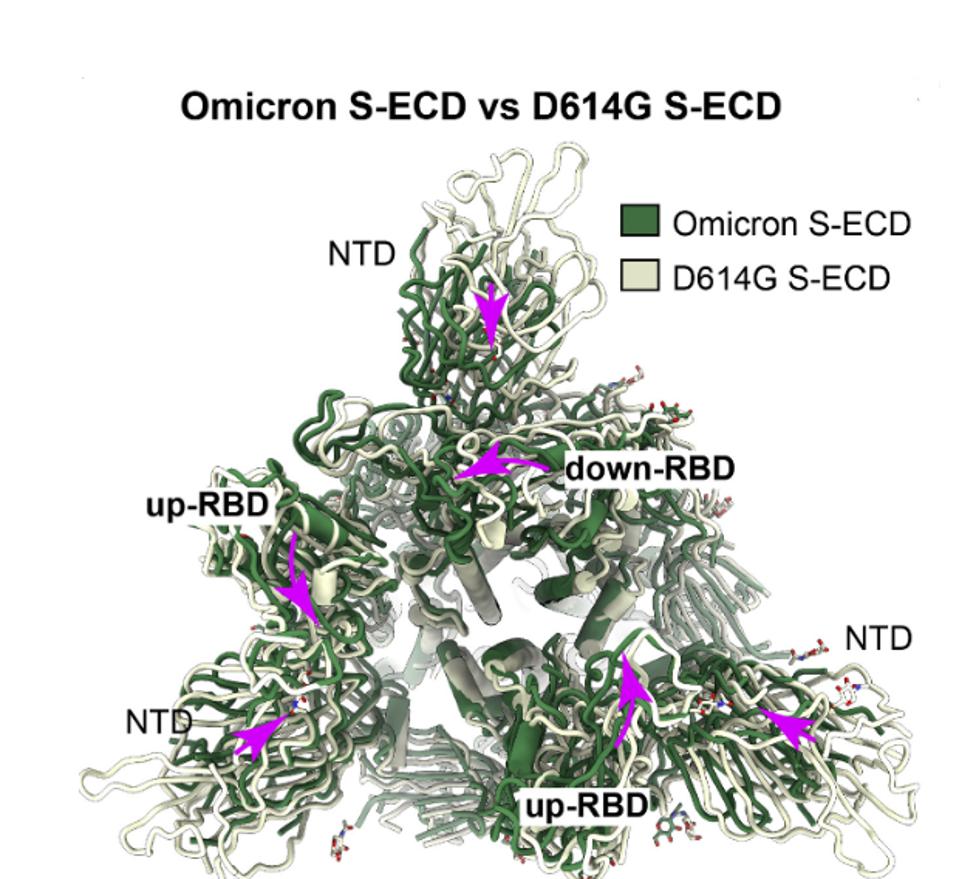
I also speculate that tighter packing implies a more entropic structure, representing a higher energy state. That additional energy may be released during membrane fusion, increasing the efficiency of viral entry.
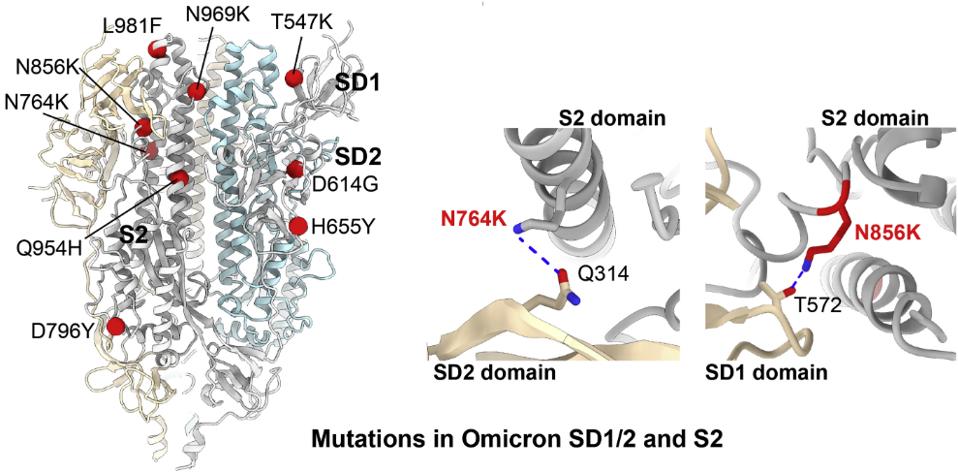
Wang et al. attribute tighter packing of the Omicron Spike to specific amino acid substitutions. The H655Y mutation induces a tighter association of the monomers that comprise the trimer. Additionally, five mutations in the central helical region, N764K, D796Y, N856K, L981F, and N969K introduce and facilitate additional hydrogen bond and hydrophobic interactions between the S2 trimers (Figure 4).
Alterations In Omicron Fusion
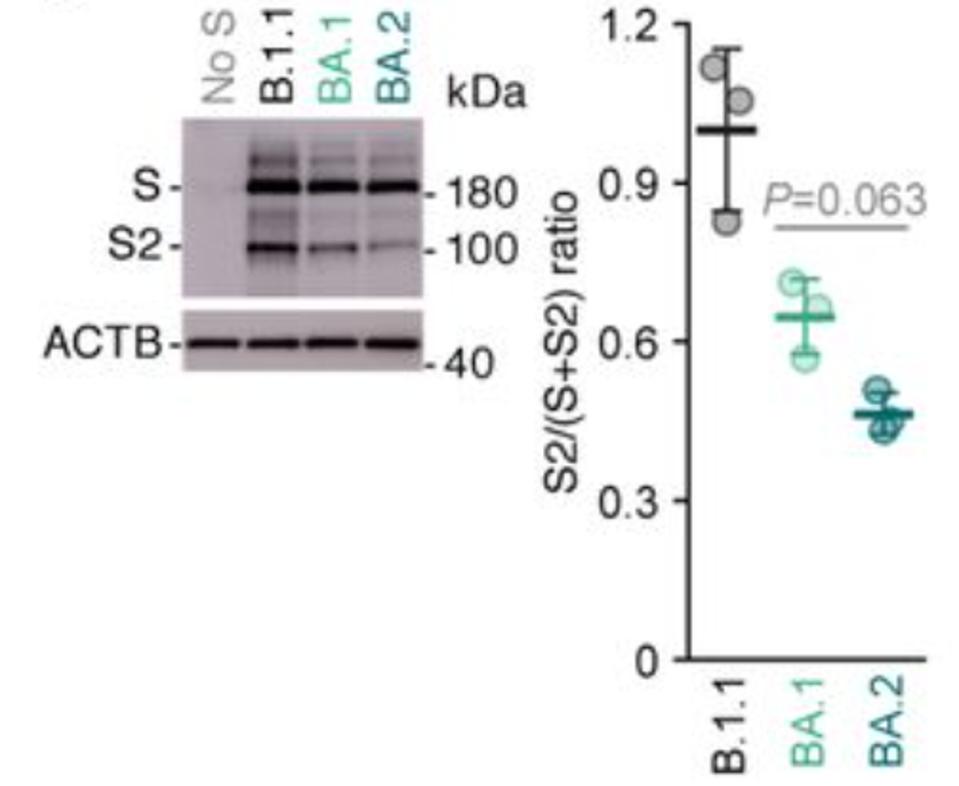
The second major structural change involves one of the hallmarks of SARS-CoV-2 as compared to SARS-CoV-1: initial scission at the S1 furin cleavage site. For most variants, furin cleaves the SARS-CoV-2 Spike protein as the virus buds from the cell surface. No such cleavage occurs for the Omicron variants, or if it does, the efficiency is greatly reduced (Figure 5 by Yamasoba et al.).
The furin cleaved S1/S2 complex is inherently less stable than the uncleaved monomer. Post-cleavage S1 readily disassociates from S2. In fact, one advantage the D614G mutant virus has over the original Wuhan variant is that it stabilizes S1/S2 association. Increased transmission of Omicron as compared to other variants may be partially attributed to increased retention of S1 on the mature virus particle.
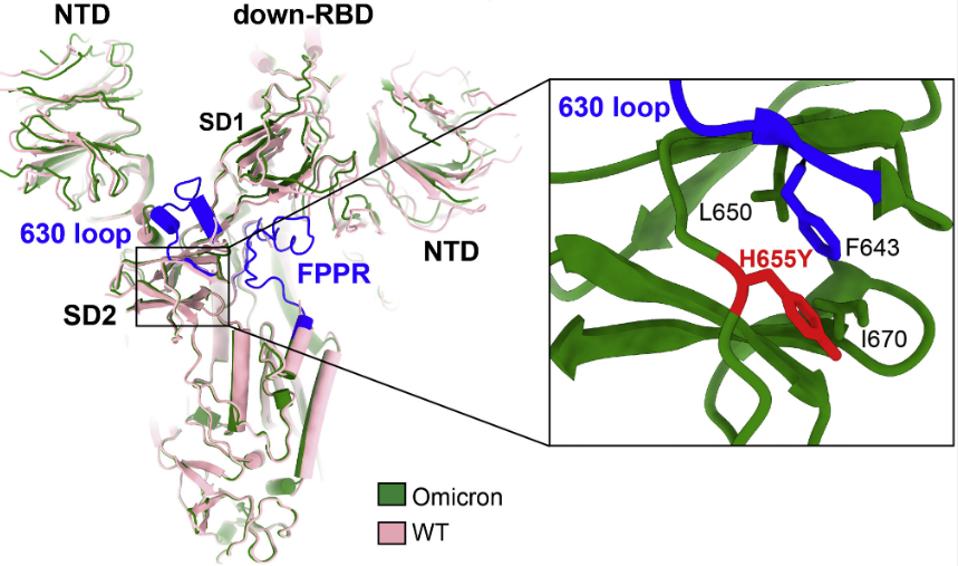
Wang et al. note that one mutation in the Omicron Spike, H655Y, may account for reduced furin cleavage at the furin cleavage site. The mutation increases the stability of the 630 loop in the region by interacting with residue F643. Wang et al. and Bing Chen speculate that increased rigidity of the loop containing the furin cleavage site reduces proteolysis.
The Wuhan strain and all previous variants enter via membrane-to-membrane fusion. The Omicron family by contrast enters via an endosomal entry route. This is the strategy employed by SARS-CoV-1. Endosomal entry is very likely required in the absence of efficient furin cleavage. The S1/S2 cleavage site is at positions 685/686 and the S2 site is at positions 815/816.

This observation prompts a paradox. The presence of the furin cleavage site was hypothesized to be a key event in the acquisition of efficient human-to-human transmission. In fact, such speculation was so prominent that some suggested the furin cleavage site was artificially inserted to increase the infectivity of the virus as part of a laboratory gain-of-function experiment. Many who made that speculation appear to have been ignorant to the fact that many naturally circulating alpha and beta coronaviruses contain furin cleavage sites It is notable that even though the Omicron family of viruses contain furin cleavage sites, they are poorly active at best.
The paradox: The Omicron family of viruses are far more infectious than previous variants. It is evident that efficient cleavage is not necessary, either for infection or efficient transmission in the human population.