
This is the eleventh article in a series called “How SARS-CoV-2 Evades And Suppresses The Immune System,” which will explore an underappreciated but highly significant aspect of SARS-CoV-2 replication. The ability of SARS-CoV-2 to delay, evade, and suppress the immune system has myriad implications for drugs, vaccines, and other aspects of our pandemic response. The first set of pieces in this series are intended for a general audience; the second set, for the medical community; and the third and final set, for biomedical researchers looking for a deeper understanding of variants, how they’re generated, and what we might do to control them. Read parts 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10.
Targeting key players in interferon induction
Discussions of innate immunity typically focus on the exterior—what happens outside the virus. But another way of approach is to focus, once the virus gets in, on the interior of the infected cell and its neighbors.
In the previous few installments of this series I discussed nonspecific mechanisms of SARS-CoV-2 immune suppression, which affect entire cellular processes and structures. Now I will shift our attention towards specific immune suppression. In addition to mounting a more targeted offensive, this component of the virus’ strategy yields very precise effects, in some cases limited to just one element of one immunological pathway. But the consequences of these minute shifts, as we shall soon see, can be far-reaching.
For example, many of the Orfs (open reading frames), as far as we know, only interact with select innate immune signals, like interferon and interferon-responsive genes, while the nonstructural protein Nsp1, in addition to its nonspecific functions, also specifically inhibits RIG-I, a pattern recognition receptor critical to the activation of the innate immune response.
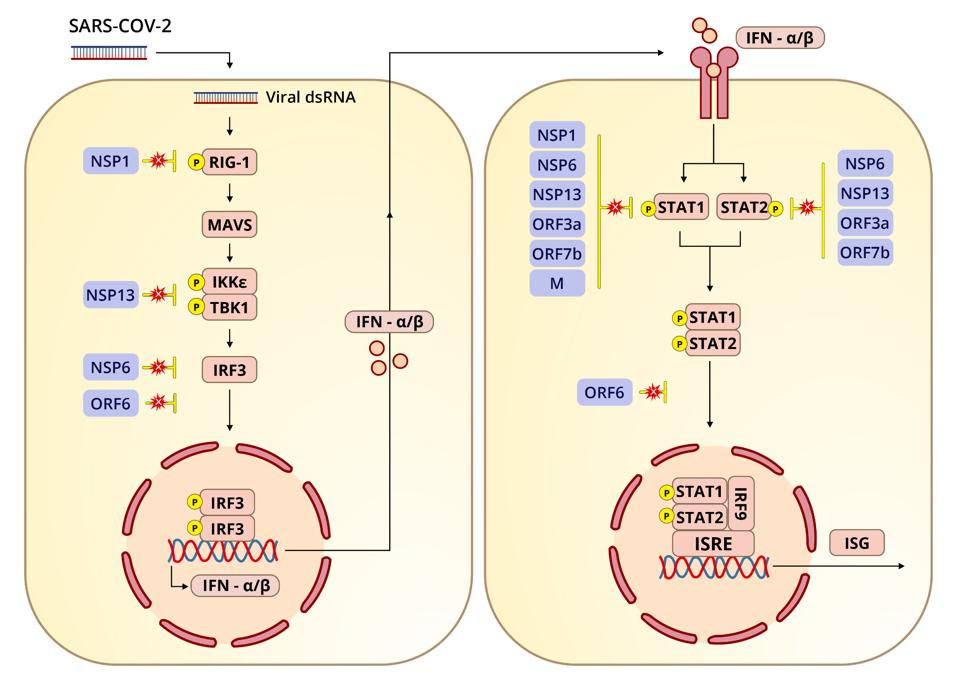
The virus devotes a tremendous array of proteins to blocking not only the induction of interferon and its export but also interferon-stimulated genes. Interferon-orchestrated innate immunity must be overcome for the virus to be successful. Many of the viral proteins are devoted precisely to this task: nonstructural, structural, and a whole set of specific proteins we call regulatory or accessory genes. Here we look at the first half of their blockage of interferon synthesis (see left panel of Figure 1).
Clinical data shows more adverse health outcomes for Covid-19 patients who either inherit genetic deficiencies in interferon or develop autoantibodies against interferon. If the body’s ability to produce interferon is compromised, it will be in serious trouble.
This is also very important for the virus’ overall masking of infection. If interferon is not induced, cells won’t be able to alert the immune system to the presence of a pathogen. One of the primary tricks SARS-CoV-2 has for expedited entry and exit is blocking not just interferon itself, but multiple steps along the interferon signaling pathway.
ACCESS HEALTH INTERNATIONAL
First, a bit of context on the significance of RIG-I. Innate immunity involves proteins known as pattern-recognition receptors that, over the course of evolution, have been hard-wired to identify pathogen-associated molecular patterns or PAMPs. Commonly found in many pathogens, recognition of these molecules allows the body to respond immediately to invaders old and new. Some of the better-known PAMPs include bacterial lipopolysaccharides, acids, bacterial DNA, and both single- and double-stranded RNA. Activation of these pathways by pattern recognition receptors like retinoic acid-inducible gene I (RIG-I)-like receptors and Toll-like receptors is necessary to trigger the adaptive immune response.
RIG-I is one of the pattern recognition receptors that detects PAMPs. The primary function of RIG-I is to recognize and bind to double stranded RNA, the key intermediates in SARS-CoV-2 replication and transcription. Upon binding to double stranded RNA, RIG-I activates mitochondrial antiviral adaptor protein (MAVS), which in turn sets off a cascade of downstream factors: k-B kinase ε (IKKε), TANK binding kinase 1 (TBK1), and interferon regulatory factor 3 (IRF3). If phosphorylated, IRF3 translocates to the nucleus and initiates transcription of type-I interferons.
One study, published in October 2020, identified four viral proteins in SARS-CoV-2 that obstruct the RIG-I pathway and therefore interferon production: the nonstructural proteins Nsp1, Nsp6, and Nsp13 and the open reading frame Orf6. If the RIG-I pathway is activated, Nsp6 and Nsp13 bind to TBK1 to halt the phosphorylation of IRF3. And if IRF3 is phosphorylated, Orf6 interferes by blocking the translocation of IRF3 to the nucleus.
Let’s dive deeper into these various points of interference, beginning with Nsp6 and Nsp13. If the RIG-I pathway is functioning as it should, IKKε and TBK1 phosphorylate IRF3. Phosphorylation, or the addition of a phosphate group, is a biochemical reaction that regulates proteins, playing a critical role in glycolysis and other activities essential to maintaining a healthy cellular metabolism. When Nsp6 binds to TBK1, the authors of the study found, the interaction inhibits phosphorylation of IRF3. When Nsp13 binds to TBK1, it reduces TBK1 phosphorylation and IRF3 activation.
Normally, once IRF3 is phosphorylated, it is translocated to the nucleus, where interferon and interferon-stimulated genes are then transcribed.
Simultaneously, the transport receptors karyopherin α 1–6 (KPNA1–6) import transcription factors that facilitate translocation, including IRF3. Karyopherins are proteins that help with the transport of molecules between the cytoplasm and nucleus by shuttling their cargo through the nuclear pore complex. They regulate what goes in and out, with different karyopherins assuming different gatekeeping tasks. But according to the October 2020 study, Orf6 inhibits translocation by binding to KPNA2. The net result, as is the case with suppression mediated by Nsp6 and Nsp13, is less production of interferon beta.
The role of Nsp1 in interferon blockage has been studied more extensively, not just in SARS-CoV-2 but SARS-CoV and MERS-CoV. The Nsp1 of SARS-CoV-2, as well as Nsp6, is more efficient at suppressing interferon production than its counterparts in SARS-CoV and MERS-CoV. In a previous installment of this series I discussed how Nsp1 broadly inhibits the synthesis of cellular proteins by binding to the 40s subunit of the ribosome. According to another study, the downstream effects of this shutdown likely reach the interferon signaling pathway. For my next piece I’ll explain how Nsp1 and a host of other viral proteins block STAT1 and STAT2 (signal transducer and activator transcription proteins).