This is a series on the advances in CAR T, a remarkable immunotherapy treatment dubbed a “living drug.” This first installment will lay the foundation for understanding how CAR T works. Future installments will focus on CAR T applications and recent innovations which further the field.
One of the dreams of cancer therapy is to use the power of the body to heal itself. This dream, long in the making, is becoming a reality thanks to deep and fundamental understandings of the immune system, the primary means by which we protect ourselves from external and internal threats. The immune system recognizes and eliminates threats, whether from viruses and bacteria from outside the body or by cells behaving abnormally within the body. Using the body’s own immune cells as anticancer agents has long been part of this dream. I was an early pioneer in creating one of the first proven cell therapies using dendritic macrophages to treat prostate cancer. Today, immune cell therapy offers hope to people with cancer and other previously untreatable diseases.
This series will explain a recent and revolutionary cell therapy called CAR T, delving into current successes and future opportunities.
The “T’ of CAR “T”
CAR T is short for Chimeric Antigen Receptor T cells. Essential to understanding this therapy is an understanding of T cells and cell-mediated immunity.
Adaptive Immunity
Adaptive immunity allows humans to form a tailored defense to foreign invaders. Adaptive immune cells memorize the telltale signs of enemies and trigger defensive mechanisms if the signs are detected in the future. This branch of immunity concerts two separate arms—humoral immunity driven by antibody-producing B cells, and cell-mediated immunity driven by “helper” T cells and “killer” T cells.
CAR T technology alters the typical functioning of cytotoxic cells. Instead of indirectly aiding antiviral processes as CD4+ helper T cells do, CAR T borrows the cytotoxic power of CD8+ killer T cells to destroy infected or abnormal host cells, thus transforming into a “living drug.” A typical cytotoxic T cell eliminates threats using the following process:
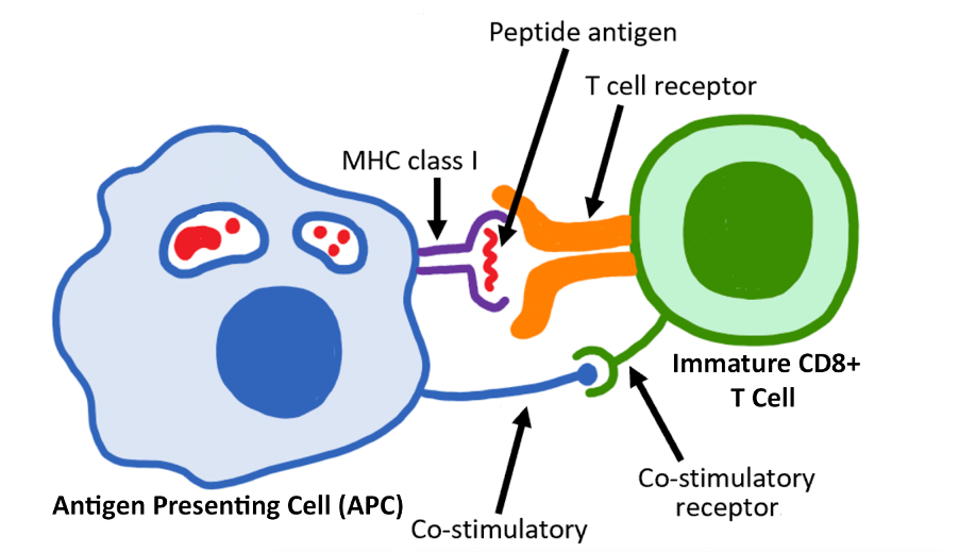
FIGURE 1: A T cell activates once it encounters an antigen presenting cell (APC) with its corresponding antigen. An antigen presenting cell (APC) breaks down foreign proteins into smaller fragments within the cell. In the case of killer T cells, a major histocompatibility complex sits on the cell surface of the APC and presents the antigen. The T cell receptor binds to the antigen; simultaneously, a co-stimulator signal is received. The result is an activated T cell. WIKIPEDIA Link Added
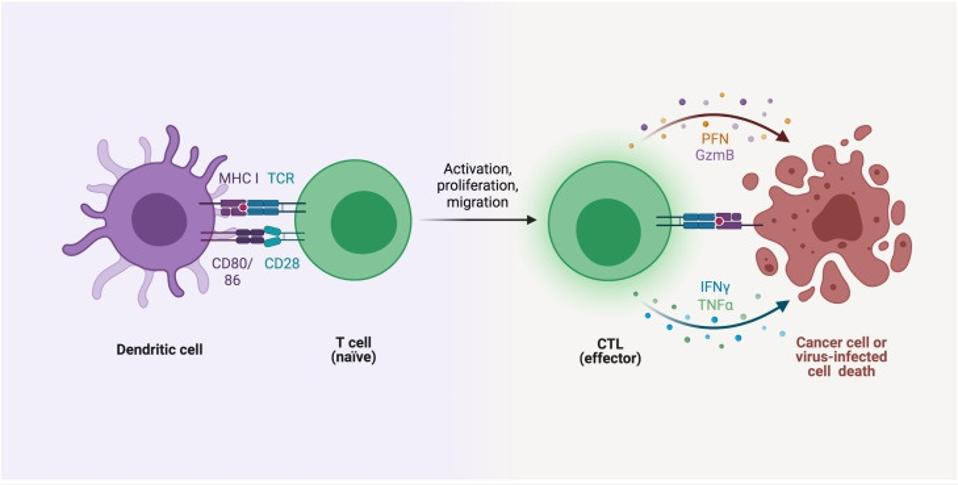
FIGURE 2: Cytotoxic T cell activation and subsequent apoptosis of cancer/infected cell SADEGHALVAD ET AL Link Added
The process (as illustrated in Figure 1) begins with the differentiation of an inactive T cell—in essence, any T cell without a specified purpose. As if waiting for the right key, a T cell does not activate unless it encounters an antigen presenting cell (APC) with its corresponding antigen. In order for an interaction between the two cells to occur, several steps must occur.
Firstly, the antigen presenting cell must process the antigen, the enemy components, into smaller peptides. Then, these peptides must be carried to the antigen presenting cell’s surface by major histocompatibility complexes (MHC). Immature CD8+ T cells require MHC Class I molecules to facilitate this translocation. Around this time, a secondary signal such as CD80 or CD86 must also be received by the T cell. In the final step, the antigen presenting cell releases a protein signal called CD40 and cooperates with helper T cells to finalize the differentiation process.
How Killer T Cells Kill
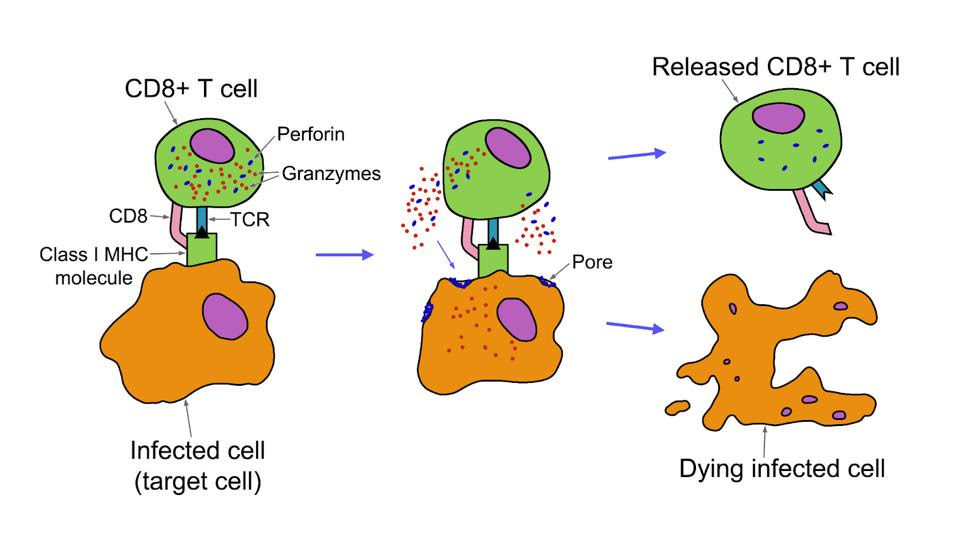
FIGURE 3: CD8+ T cell induces apoptosis of a target cell. DANANGUYEN DERIVATIVE: NAGUALDESIGN
Killer T cells destroy infected and abnormal cells by inducing apoptosis, a form of controlled cell death which does not spark inflammation. Pockets of enzyme within the T cell must make contact with the target cell to trigger its death.
When a cytotoxic T cell recognizes its target, it binds to the class I MHC molecule on the surface of the target cell (see Figure 3) to create a bridge. With the bridge completed, the T cell can then release the enzymes. One enzyme drills pores in the target cell’s membrane, thus ruining its integrity. The other travels through these newly made tunnels, tipping an enzyme cascade inside the target cell which accelerates its degradation.
The crumbling target cell mimics the imagery of bricks falling from castle walls. Nearby phagocytes recognize the “crash of bricks”—more accurately, sense a change in membrane—and begin ingesting the target cell. The target cell breaks down to nothing inside the phagocyte without stimulating inflammation or other side effects.
Construction of a Chimeric Antigen Receptor
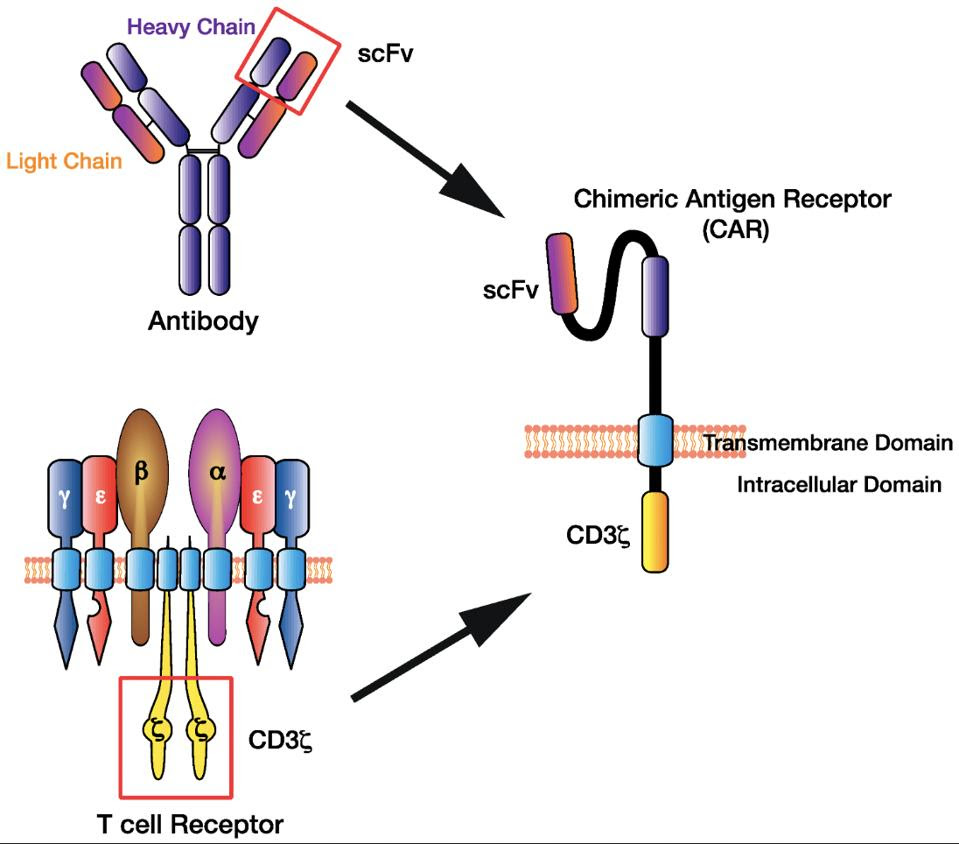
FIGURE 4: The basic design of a chimeric antigen receptor (CAR) synthetically combines the structure and function of T cells and B cells. It borrows the single chain variable fragment (scFv) from antibodies and the CD3 co-receptor from T cells. The result is a cytotoxic T cell with the targeting power of antibodies. This design can be altered further to increase precision targeting. CREDIT: HUGHES-PARRY ET AL Link Added

FIGURE 5: Modifications to CAR design. Many of the changes improve antigen targetability, CAR T cell function and applicability. Abbreviations: dual chain CAR (dcCAR), the T cell receptor fusion construct (TRuC). CREDIT: HUGHES-PARRY ET AL Link Added
Killer T cells are clearly useful in clearing irregular host cells. Researchers recognized this and sought to harness this natural design to eliminate cancer cells through CAR T. Chimeric antigen receptors are engineered to detect a specific antigen and trigger the destruction of a target cell. The most basic CAR T design accomplishes this by manipulating antigen binding sites normally intrinsic to antibodies—single chain variable fragments (scFV)—to lend cytotoxic T cells higher antigen specificity. The CAR T cell recognizes specific antigens thanks to this domain.
Next comes the flexible hinge region. This region simultaneously stabilizes the CAR while its length provides grants easier access to specific antigens. The transmembrane domain anchors the antibody and hinge structure.
The intracellular domain describes receptors lying within the T cell. Basic CAR T design employs CD3 here, a T cell receptor needed for T cell differentiation (see Figure 4). Second and third generation CAR models included secondary signal receptors such as CD28 to improve target cell elimination and cell signaling (Figure 5).
More recent research developments in CAR design deviate from this foundational model to finetune precision and function. For example, T cell receptor fusion construct (TRuC) CAR tethers the scFV region to the several intracellular CD3 subunits, thereby reducing secondary signaling hypothesized to be unnecessary. Universal CAR (uCAR), on the other hand, augments antibody specification by fusing biotin to the transmembrane domain and the endodomain. Other research efforts incorporate cytokines (signaling molecules) and other molecules to improve T cell expansion and persistence, as well as synthetic control switches to minimize the therapy’s toxic side effects. The groundwork model inspires many alternative CAR designs beyond those demonstrated here.
The beauty of this science lies in the melding of two previously separate abilities. CAR T therapy replaces the T cell receptor with an antibody-like structure, all while maintaining the transduction machinery of a T cell. Like this, MHC class I binding becomes irrelevant and a response can be immediately triggered.
The CAR T Therapy Process
What does the CAR T therapy process look like?
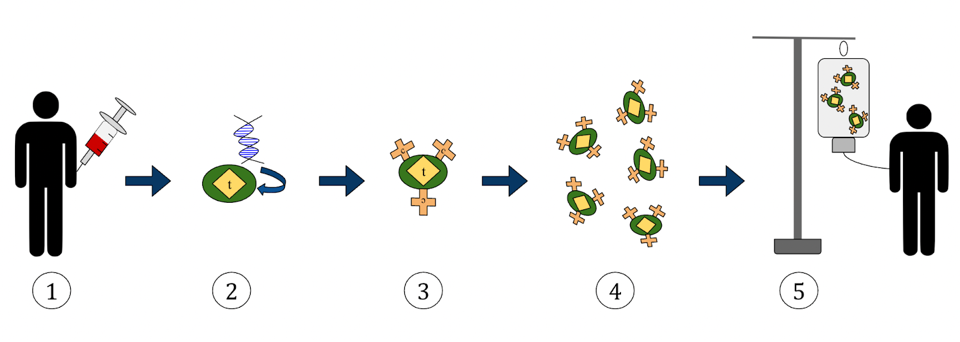
FIGURE 2: Chimeric Antigen Receptor T Cell production and infusion, broken down into five steps, as follows: (1) Isolation of T cells (2) Incorporation of a gene encoding chimeric antigen receptor in the T cells (3) T cells gain a specific target antigen (4) Engineered T cells proliferate in cell culture, and (5) Infusion of engineered cells into patient. WIKIPEDIA Link Added
Figure 2 illustrates the progression clearly. For a patient receiving CAR T therapy, the process may begin with a medical professional drawing their blood and separating T cells from that sample using apheresis; this would be an autologous treatment, as the cells used originate from the same patient. T cells can also be isolated from a healthy donor’s blood sample, otherwise known as allogeneic transplantation.
The cells must then be genetically altered to recognize a particular target in a cell processing center. To do this, the cells are “expanded”—a process which stimulates T cell proliferation. The new plethora of T cells must be purified and then genetically modified with a gene that encodes the desired chimeric antigen receptor. CRISPR technology can be used here to accomplish the task.
The cells are now ready for infusion. The cells are frozen and sent back to the treatment center. The patient preps for infusion with a lymphocyte-depleting chemotherapy; the chemotherapy reduces the number of white blood cells in the blood to reduce competition for the CAR T cells, thus helping them multiply. With success, the engineered T cells will recognize the antigen on cancerous cells, bind to it, and mark it for destruction via apoptosis. The infusion takes between 30 to 90 minutes to complete, but the patient will be closely monitored for days, weeks or months to watch for any adverse side effects and to receive additional treatments.
Side effects can occur if the “living drug” multiplies too actively, the most common being cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). Also commonly known as “cytokine storm,” CRS occurs when proteins called cytokines flood the immune system and send it into overdrive. Symptoms tend to be mild—fever, nausea, headache, rash, and more—and resolve within a couple of days, but they can also be severe or life threatening. ICANS refers to a neurotoxic condition that appears within one to three weeks after T cell infusion. Early signs, such as tremor and lethargy, can lapse into stupor, seizures or coma if untreated. More on managing side effects to come in later installments in this series.
The long term side effects of CAR T are unknown. As a result, the FDA stipulates that gene editing treatments such as CAR T therapy should be monitored for up to 15 years—five years of annual follow ups, followed by ten years of questionnaires and/or other queries.
What Illnesses Can CAR T Treat?
CAR T therapy is FDA approved to treat B cell-derived lymphomas—cancers caused when B cells (not T cells) grow too rapidly—as well as multiple myeloma, cancer of plasma cells found in the bone marrow. These treatments tailor chimeric antigen receptors to target an antigen called CD19 found only on the tumor cells of lymphoma patients. Another target is BCMA, a B cell maturation antigen specific to multiple myeloma.
CAR T therapies may be federally approved, but they are not used as first or second line cancer treatments; usually CAR T therapy is considered after receiving standard chemotherapy treatment and other alternatives. And as a newer treatment, it may be more expensive than other therapies or may not be fully covered by health insurance.
But this field is ever growing. Several hundred clinical trials are in progress to test the boundaries of this mechanism and enhance its design. The next installations in this series will cover some of the most recent discoveries in the CAR T circuit, such as treatment advances in B cell lymphomas, lupus and heart disease, as well as innovations in CAR T precision.