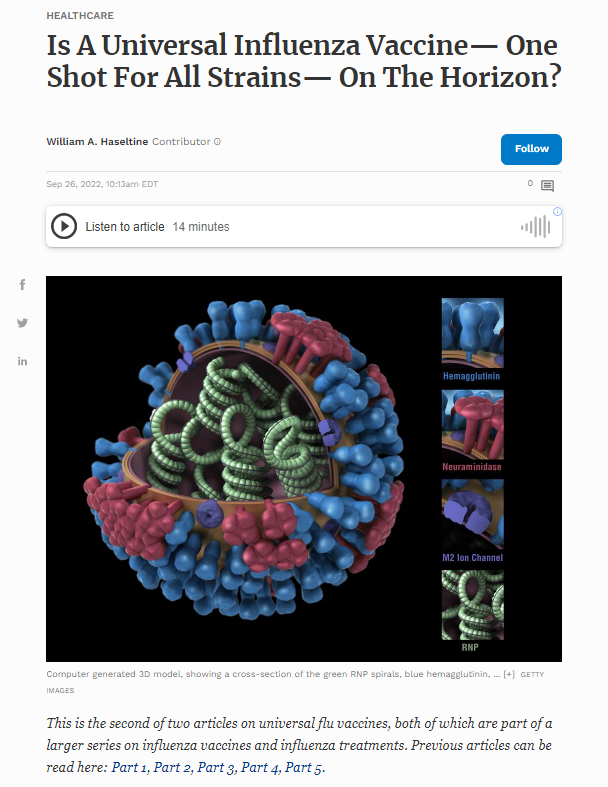
This is the second of two articles on universal flu vaccines, both of which are part of a larger series on influenza vaccines and influenza treatments. Previous articles can be read here: Part 1, Part 2, Part 3, Part 4, Part 5.
For many years there has been an obvious need for an influenza vaccine that neutralizes not just one strain, but many different strains across both major influenza types. This includes providing protection against as-yet undiscovered strains. Current vaccines have not achieved this, requiring yearly administration to keep pace with viral mutations. But over the last decade there has been great effort to develop such a universal flu vaccine, with much of it now beginning to pay off.
Two Targets Are Better Than One
The key problem so far has been that flu vaccines provide a potent but highly specific immune response; they work decently well, but only against those strains that have been included in the vaccine. Should a new strain of influenza appear, or one that scientists hadn’t predicted, then vaccine efficacy is left seriously jeopardized. To combat this issue, scientists have begun looking towards highly conserved regions of the viral genome. These are regions that are shared across many strains and that only very rarely mutate.
Two areas of the influenza virus particle are particularly promising targets: the stem portion of the influenza surface protein hemagglutinin (HA), and the extracellular region of the matrix-2 protein (M2e). Both of these proteins play a crucial role in viral replication — M2 helps untether the viral RNA from the surrounding viral membrane, and HA stem (HA2) creates the opening through which the RNA can then enter the host cell cytoplasm. Antibodies that bind to and block either of these proteins impair viral replication. Here, we describe a new vaccine candidate developed by Subbiah et al. that combines these two attractive targets into a single, highly engineered protein.
Design of the Antigen: Perfecting the “Headless” HA Protein
Interest in the stem portion of HA has its roots in the discovery of human broadly neutralizing antibodies (bnAbs) that target the region. This discovery revitalized hopes of developing universal flu vaccines by giving researchers an entry point for the production of a universal antigen. Since the full HA protein only rarely elicits broadly neutralizing antibodies, various approaches have emerged to expose and properly present the stem domain to our immune system. One of the more successful strategies involves knocking off the HA head domain while ensuring structural stability of the leftover HA stem.
But this is easier said than done. The head of the HA protein stabilizes the prefusion protein resulting in a tightly coiled triplex of three proteins. Broadly neutralizing monoclonal antibodies only recognize this tightly coiled triplex and are lost once the fusion process commences or the head of the HA protein is removed.
To perfect the hemagglutinin portion of their antigen, Subbiah and colleagues drew upon previous research by Impagliazzo et al. and Yassine et al. Both groups managed to engineer stable, headless HA stem antigens —called “mini-HAs”— that exhibited structural and antibody binding properties similar to full-length HA. Antibodies produced in response to these mini-HAs managed to protect against a wide range of HAs in mice and nonhuman primates.
Figure 1 outlines the various stages of Impagliazzo et al.’s design, culminating in the creation of a stable, headless “mini-HA” stem that retains all the relevant neutralizing binding sites. Figure 2 outlines the design stages of Yassine et al, who achieved great minimization of the HA protein, ultimately succeeding in having the stem domain contribute 94% of the total surface area of their antigen.
Forming a Stable Trimer and Preventing Protein Aggregation
Influenza hemagglutinin is naturally a trimeric molecule, meaning it is composed of three distinct hemagglutinin proteins bound together. In order to accurately mimic this three-part structure, Subbiah et al. excluded the C-terminus of the HA stem portion and instead replaced it with the foldon sequence of a virus that infects bacteria.
When added to the end of a monomer that has a propensity to dissociate, the foldon sequence stabilizes it and allows it to form a trimer (Figure 3). Failure to do so may lead the headless HA stem antigen to lose its structure, and by extension its epitope sites. If our antibodies cannot bind to the stem, they cannot learn about its structure and thus cannot take on the specific shape needed to protect us from future reinfection.
The researchers also added specific point mutations along HA1 head and HA2 stalk to prevent protein aggregation issues. All of the mutations were in regions outside of those recognized by broadly neutralizing antibodies. Each one of the mutations was in a long helical region with a tendency to self-aggregate. By changing the amino acids in these stretches, they substantially reduced non-specific aggregation and instead favored the stable form of the protein.
In addition to this, they mutated a cysteine residue and replaced it with a serine residue. Again strengthening the stability of the protein and minimizing the chance of any unwanted disulfide bonds from forming.
Addition of M2e
To broaden the neutralization of their antigen, and increase its potency, the researchers added in two identical segments of the M2 channel protein. The segments they chose correspond to a 23 amino acid-long sequence that encodes the epitopes for the extracellular region of the protein. This area protrudes from the viral membrane, allowing it to be bound by neutralizing antibodies.
Crucially, M2e has previously been shown to confer cross-group immunity. Influenza A viruses are split up into two groups according to phylogenetic differences in their HA protein. Usually immunity against group 1 HAs (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, H18) does not confer immunity against group 2 HAs (H3, H4, H7, H10, H14, H15). This means that infection by an influenza A/(H3N2) virus, for example, generally does not confer protection against a strain of influenza A/(H1N1).
Finally, Subbiah et al. connected the various domains of the M2e-H3 construct using flexible linkers, which are small amino acids that function like bungee cords; they keep the proteins tethered together but are flexible enough not to interfere with the folding process.
The full genetic sequence of the M2e-H3 antigen can be seen in Figure 4, and the structure of the antigen —as compared to wild type hemagglutinin— can be seen in Figure 5.
Production and Purification of the Antigen
For ease of purification of the antigen, the researchers added a histidine tag (6xHis) to the N-terminal of the M2e-H3 stem domain. A histidine tag is made up of a DNA sequence that encodes six to nine histidine residues. These histidine residues bind to several different types of immobilized metal ions. So, once a protein has been “His-tagged” it can be easily identified and separated from other proteins by exposure to such metal ions. The histidine tag can then be removed from the protein or it can be retained, either way it does not affect the antigenicity of the protein.
Next, Subbiah et al. synthesized a codon-optimized gene encoding the M2e-H3 stem protein. This gene is then inserted into escherichia coli (E.coli) for quick and easy mass production.
Does it Retain Antigenicity?
After extraction and purification, Subbiah et al. began testing the M2e-H3 stalk molecule for its ability to be recognized by antibodies, known as antigenicity. They exposed the chimeric molecule to antibodies known to target the stem portion of the hemagglutinin protein as well as antibodies known to target M2e; in both cases they witnessed high reactivity. The scientists recorded similar results from antisera — blood samples from mice that had previously been infected with an influenza virus and had built up a corresponding antibody response. Importantly, the construct retained its antigenicity when exposed to influenza A viruses belonging to different HA groups. And, even after storage at 122 degrees Fahrenheit for 11 days, the antigen continued to elicit strong antibody reactivity, suggesting solid thermostability.
Animal Models
Having confirmed that the M2E-H3 stem protein can be recognized and bound by antibodies, Subbiah and colleagues moved on to vaccination trials. Often, vaccines based on viral proteins instead of inactivated or attenuated viruses require an adjuvant to help stimulate a stronger immune response, and by extension, better protective efficacy. For their experiments, the researchers used a particularly strong adjuvant similar to the AS01 liposome adjuvant licensed for use in herpes Zoster vaccination. This is a double-edged sword: the adjuvant ensures a quick immune response, but because of its strength, it also comes at the cost of side effects. Although the side effects are not much cause for concern —headache, fever, chills, and so on— they are uncomfortable all the same. Using a different delivery platform, like mRNA technology, may help circumvent these issues in the future.
The adjuvanted M2e-H3 stalk vaccine was administered to mice as two doses, a primary vaccination followed by a boost two weeks later. Following the first dose, there was a noticeable increase in antibodies specific to M2e and HA stem regions. Addition of the boost further increased antibody titers 10 fold. Antisera built up in response to M2e-H3 exposure managed to protect against both group 1 and group 2 influenza A viruses. Subbiah et al. suspect this might be explained by antibody-dependent cellular cytotoxicity (ADCC), a mechanism of immune defense whereby effector cells recognize antibodies stuck to antigens and actively break down the foreign threat. Their suspicions were proven right by western blotting, which showed that M2e antibodies, in particular, raised ADCC activity 20 fold.
To compare protection against an M2e only vaccine, the group of researchers designed another chimera protein combining M2e fused with the receptor binding domain (RBD) of SARS-CoV-2 spike protein. When exposed to a lethal dose of influenza A/HK/H3N2 virus, all of the mice inoculated with M2e-H3 stem survived, suffering only moderate body weight loss of around 10%. On the other hand, none of the mice inoculated with the M2e-only vaccine survived the infection (Figure 6).
Along with a robust antibody response, M2e-H3 stalk vaccination also triggered a strong T cell response. Where antibodies can bind to pathogens to prevent infection, T cells are in charge of getting rid of already-infected cells, curbing the spread of the virus. Indeed, the vaccine significantly reduced the amount of virus in the lungs of infected mice within a span of six days. Depleting T cell levels in vaccinated mice before exposing them to a lethal challenge with influenza A H7N9 produced twice as much weight loss (12%) than in mice that had not been depleted of their T cells.
Implications
The work by Subbiah et al. acts as a clear proof of concept: using M2e and HA stalk regions as antigens can stimulate broad neutralizing immunity. Crucially, this immunity should hold up to new strains, as it targets those viral regions that only very rarely mutate. Although their work focused specifically on influenza A viruses, there is no reason to think that, given enough time and research, we won’t be able to produce a truly universal influenza vaccine.
The progress that has been made is in very large part due to the development of monoclonal antibodies that are broadly neutralizing against a wide array of influenza strains. These antibodies have provided a key guide to the design of vaccine antigens to help elicit immune responses with a similar breadth of neutralizing capabilities. This elegant work that has developed over the past ten years may prove to be a reliable framework for the development of similarly universal vaccines to some of our most vexing problems, including HIV and SARS-CoV-2. Such attempts are already underway for Covid-19, and proved to be the missing part of the puzzle for the design of effective vaccines against respiratory syncytial virus (RSV).