
This article is an extension of our series on immune suppression by SARS-CoV-2. The series has since been published as a book, Natural Immunity and Covid-19: What it is and How it Can Save Your Life. It is also available to read on my website.
One of the keys to SARS-CoV-2’s rapid spread and global persistence is its ability to evade and suppress our immune system. Although other viruses also implement a variety of tactics and tricks, SARS-CoV-2 is particularly cunning. Adding to an ever-growing list, researchers at the Perelman School of Medicine at the University of Pennsylvania have recently uncovered another such SARS-CoV-2 strategy: mimicry of human histone proteins. Published in Nature, Lee et al. show that one of the virus’ accessory proteins —ORF8— mimics host histone protein H3, interfering with cell gene regulation and immune defense.
Most living cells carry chromosomes in their nucleus. These are thread-like structures made up of long, thin DNA fibers. They carry genetic information in the form of genes, which allows cells to be accurately copied during cell division. Importantly, the DNA strands are coated with packaging proteins known as histones (Figure 1). Think of these as genetic on/off switches: the more tightly packed the histones, the harder it is to read the DNA, and the more loosely packed, the easier. This allows cells to regulate gene expression, including genes related to our immune response. Combined, the DNA fibers and histone proteins are known as chromatin.
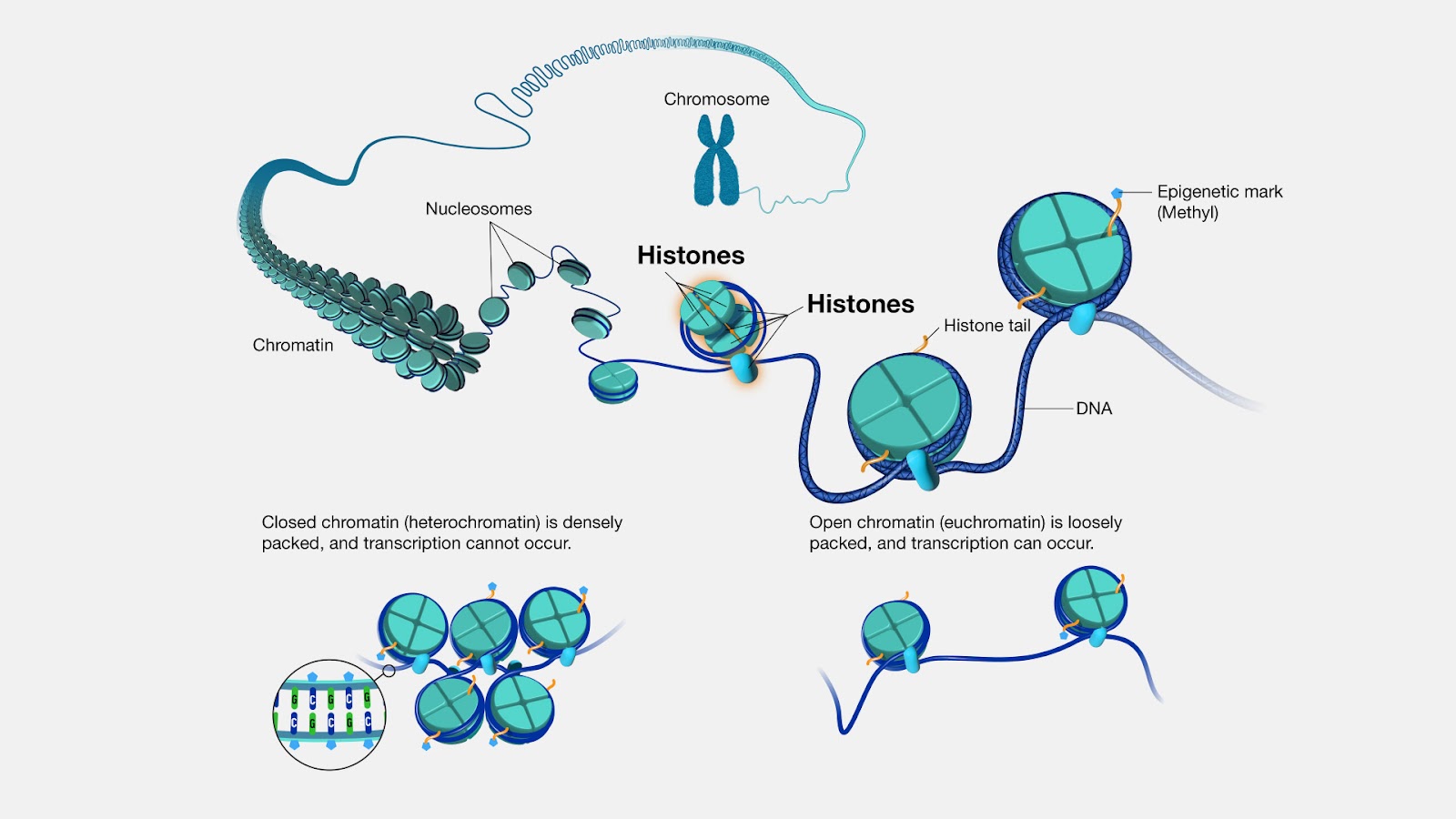
Previous research had shown that SARS-CoV-2 can impede chromatin regulation, leading to reduced cellular antiviral responses. But the exact mechanism remained poorly understood.
Lee et al. suspected histone mimicry may play a role.
Master Impersonator: ORF8 Mimics Histone H3
To investigate their hunch, the scientists performed a bioinformatic comparison of all SARS-CoV-2 proteins with all human histone proteins. They noticed that a genetic sequence of the viral accessory protein ORF8 shared a striking similarity to a sequence found in the tail region of histone H3—one of the five main histones. The culprits? Six amino-acid residues: alanine, arginine, lysine, serine, alanine and proline (Figure 2). The first four of these were found to repeat in a second site within the tail-region of histone H3.
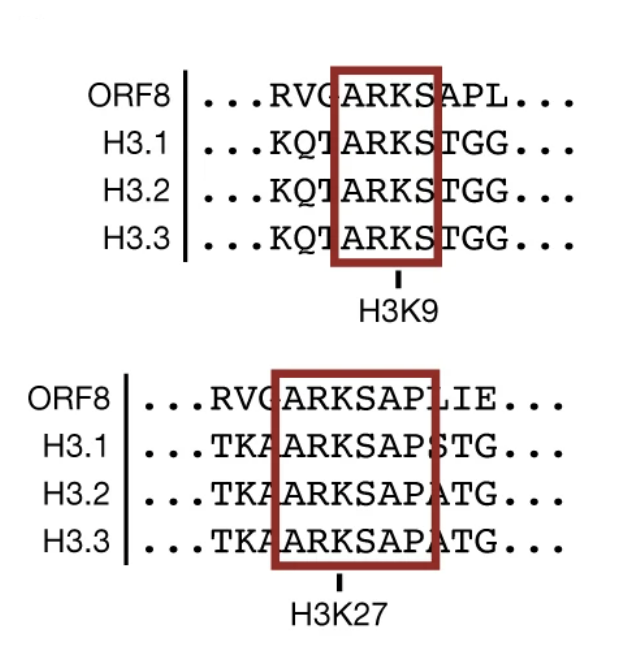
The ARKS genetic motif is well known as an important site for gene regulation within histone H3. Recall that histones act as on/off switches for gene expression. They are turned on or off by enzymes through the addition of molecular groups; acetyl groups switch the DNA “on”, methyl groups switch the DNA “off”.
During viral infection, the immune system depends on such post-translation regulation to orchestrate an effective defensive response — to modulate inflammation, recruit the appropriate immune cells, and so forth. Interfere with histone-mediated gene expression and you might interfere with antiviral responses. ORF8, by mimicking histone H3, could be doing just that.
Chromosomes are located in a cell’s nucleus. But it is rare for SARS-CoV-2 proteins, and coronavirus proteins as a whole, to be able to enter the nucleus. If ORF8 were mimicking histone H3, it would need to be able to enter the nucleus. To find out if ORF8 met this precondition, Lee and colleagues exposed human cells to ORF8. Sure enough, they discovered that the viral protein could pass into the nucleus of the cells. Once inside, ORF8 interacted with chromatin and histone H3, modifying gene expression. The same ORF8 protein with the ARKS motif removed also managed to enter the nucleus but displayed only a weakened ability to bind chromatin.
The researchers noticed that an enzyme called histone acetyltransferase KAT2A, commonly referred to as just KAT2A, interacts with the ARKS motif of ORF8. Usually this enzyme interacts with histone H3 by adding an acetyl group to the lysine amino acid in the ARKS motif — turning “on” certain genes. It was doing the same thing with the ARKS motif of ORF8. In fact, when ORF8 was added to cells, the levels of KAT2A dramatically decreased. Lee et al. speculate that ORF8 was degrading the enzyme after binding to it, preventing the enzyme from interacting with its intended target, histone H3.
The drop in KAT2A was associated with a reduction in active gene expression; the histone modifications that usually turn on these genes weren’t taking place, since ORF8 was successfully diverting traffic. Instead, histones became more tightly packed and DNA transcription —the process by which DNA is copied to RNA for the production of proteins— decreased as well.
What about SARS-CoV-2?
OFR8 in isolation was having these effects, but would it have the same effect during live SARS-CoV-2 infection? To test this, the scientists infected human cells with wild-type SARS-CoV-2. As before, ORF8 managed to interfere with histone-associated gene expression. Modified SARS-CoV-2 lacking the ORF8 gene, in contrast, was not able to disrupt transcription. A version of the virus that contained the ORF8 gene, but modified in such a way that the ARKSAP motif was missing, also failed to interfere with gene expression.
Compared to the wild-type strain, the strain lacking ORF8 entirely had a lower replication rate. Interestingly, the strain with the modified ORF8 matched the wild-type virus. This is in line with prior research indicating that ORF8 has a variety of different ways of helping SARS-CoV-2 suppress and evade immunity.
In addition to impacting replication rates, ORF8 modification also impacts transcription within the host cell: all three of the strains induced different transcriptional responses, but only the wild-type strain managed to mimic histone H3 and disrupt host-cell chromatin.
Implications
This work by Lee et al. contributes to a growing body of research on the immuno-evasive and immunosuppressive abilities of SARS-CoV-2. It should serve to remind us of two critical points.
First, although the spike protein is vital to infection —after all, it is the primary point of contact between virus and host cell— it is by no means the only relevant viral protein; SARS-CoV-2 has a myriad of accessory proteins, many of which have tangible effects on the infection process. Lest we allow the virus to get an edge on us, we need to remain well-informed about the entirety of the viral genome.
ORF8, in particular, has proven to be a swiss-army knife of SARS-CoV-2 immune obstruction. Aside from its role in histone mimicry, it also:
-
Stimulates the lysosomal degradation of major histocompatibility complex type-I (MHC-I) molecules — think of these as emergency flares that attach to the surface of cells, alerting nearby immune cells of infection. ORF8 hijacks the Beclin 1 autophagy initiation pathway, leading to autophagy of MHC-I. As a result, infected cells remain invisible to the immune system, making them less likely to be destroyed and allowing for continued viral replication.
-
Antagonizes the retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA-5) signaling pathway by targeting HSP90B1, a chaperone protein that helps fold a variety of different toll-like receptors (TLRs). This inhibits the production of type 1 interferons (IFN-I), a collection of signaling proteins that play a vital role in orchestrating immune responses. Downregulation of IFN-I helps improve viral replication during the early stages of infection.
-
Manages to escape degradation in the endoplasmic reticulum (ER) by forming disulfide complexes with resident ER proteins. The endoplasmic reticulum is where newly synthesized proteins are folded into shape; during viral infection, it is a key component of virion assembly and maturation. By forming disulfide complexes, ORF8 remodels the morphology of the endoplasmic reticulum, accelerating the transportation of proteins from the endoplasmic reticulum to the Golgi apparatus. This may help speed up virion assembly and maturation.
-
And finally, ORF8 mimics host interleukin 17 (IL-17) —a family of proinflammatory signaling proteins. ORF8 binds to the main interleukin 17 receptor, called IL-17 receptor A (IL17RA), activating the IL-17 signaling pathway and triggering inflammation. ORF8-induced inflammation is a driving force of immune cell infiltration and lung injury, including fibrosis and dysregulation of coagulation. SARS-CoV-2 variants lacking ORF8 are associated with milder infection and improved disease outcome.
This brings us to the second important implication: to effectively disrupt a viral mechanism, you first need to understand it. The more we know about SARS-CoV-2, the better we can design drugs and therapeutic interventions. By expanding our knowledge of virus-host interactions we begin to reveal areas of potential weakness, multiplying our targets.